Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Roberta Lattanzi | + 2021 word(s) | 2021 | 2021-11-21 03:35:26 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Lattanzi, R. Prokineticins and Prokineticin Receptors. Encyclopedia. Available online: https://encyclopedia.pub/entry/19622 (accessed on 28 June 2026).
Lattanzi R. Prokineticins and Prokineticin Receptors. Encyclopedia. Available at: https://encyclopedia.pub/entry/19622. Accessed June 28, 2026.
Lattanzi, Roberta. "Prokineticins and Prokineticin Receptors" Encyclopedia, https://encyclopedia.pub/entry/19622 (accessed June 28, 2026).
Lattanzi, R. (2022, February 18). Prokineticins and Prokineticin Receptors. In Encyclopedia. https://encyclopedia.pub/entry/19622
Lattanzi, Roberta. "Prokineticins and Prokineticin Receptors." Encyclopedia. Web. 18 February, 2022.
Copy Citation
Prokineticins are a new class of chemokine-like peptides involved in a wide range of biological and pathological activities. In particular, prokineticin 2 (PK2), prokineticin receptor 1 (PKR1) and prokineticin receptor 2 (PKR2) play a central role in modulating neuroinflammatory processes. PK2 and PKRs, which are physiologically expressed at very low levels, are strongly upregulated during inflammation and regulate neuronal-glial interaction.
neuroinflammation
prokineticins
GPCR
blood–brain barrier
1. Introduction
The central nervous system (CNS) has always been considered a district protected from inflammatory processes due to the presence of the blood–brain barrier (BBB). It was thought that inflammation could only be caused by the rupture of this with the consequent infiltration of immune cells. It has recently been shown that CNS inflammation is a much more complex process called “neuroinflammation”. This term indicates the activation of microglia, astrocytes, neurons and endothelial cells that induce an alteration of the permeability of the BBB. This results in increased leukocyte infiltration in the brain, increased secretion of proinflammatory cytokines and chemokines, and ultimately, neuronal damage and death [1]. Neuroinflammation plays a central role in the development and progression of many neurodegenerative diseases such as Parkinson’s disease, psychiatric and behavioral disorders, multiple sclerosis, and Alzheimer’s disease.
During neuroinflammation, to the three main occurring processes represented by the weakening of synapses, the inhibition of neurogenesis and the death of neurons actively participate those who are now considered the main actors of neuroinflammation: neurons, microglia and astrocytes. Indeed, it has been definitively established that neurons, traditionally thought to be passive bystanders in neuroinflammation, are capable of producing various mediators, such as prostanoids, cytokines (such as IL-1β, IL-6 and TNF-α) and inducible enzymes (iNOS) [2]. Also microglia are a central player in neuroinflammation and interact with other cell types that regulate neurodegeneration. Depending on the acquired phenotype, microglial cells may play a positive or negative role in neuronal survival. Inflammatory microglia exhibit the M1 phenotype and stimulate astrocyte activation, neuronal damage, T cell activation, and BBB disruption, all effects that promote neuroinflammation and subsequent neuronal loss. Conversely, microglia exhibiting an M2-type anti-inflammatory phenotype attenuate the inflammation and neurotoxic effects induced by M1-type microglia by supporting neuron survival, limiting barrier damage, and promoting repair of damaged tissue [3].
Finally, under homeostatic conditions, astrocytes have been demonstrated to play a role in several crucial biological processes. They support BBB endothelial cells, help maintain ionic balance and pH, contribute to synaptogenesis, modulate information processing and signal transduction, regulate synaptic and neuronal plasticity, deliver nutrients to neuro-glial cells, maintain excitability and neuronal network connectivity [4].
Conversely, during neuroinflammation, astrocytes become activated following stimulation by pro-inflammatory mediators, which, depending on the phenotype acquired, can affect any of these highly regulated processes. In LPS-induced neuroinflammation astrocytes, acquire an A1 neurotoxic phenotype loosing normal function, such as the ability to induce synapse formation. Other insults, such as ischemia, can induce a protective A2 reactive astrocyte to secrete neuroprotective molecules that promote CNS recovery and repair. Importantly, recent transcriptome analysis studies have demonstrated that reactive astrocytes can exhibit a gradient of phenotypes akin to activated microglia and T lymphocytes. As such, it is not possible to make a clear distinction between the two previously established activation states (A1 vs. A2) [5].
Prokineticins belong to a family of small secretory proteins that are highly conserved in evolution from invertebrates to humans. The first member of this family to be isolated was a nontoxic component of black mamba snake venom called Mamba Intestinal Toxin 1 (MIT-1) because it caused contraction of the guinea pig ileum [6]. In the same years, a small protein of 77 amino acids was isolated from the skin secretions of the frog Bombina variegata and named Bv8 [6]. Later, two human Bv8-like proteins were identified and named prokineticin 1 (PK1, or EG-VEGF) and prokineticin 2 (PK2, or mammalian Bv8) based on their ability to trigger contraction of the guinea pig ileum [6]. These proteins share some structural features: an amino-terminal AVITGA peptide segment important for biological activity and receptor recognition, ten cysteine residues forming five disulfide bridges, and a tryptophan residue at position 24 essential for binding to prokineticin receptors [7][8]. These are two related G protein-coupled receptors (PKR1 and PKR2) that binds PK1 and PK2 with the same affinity. Prokineticin receptors couple to Gas, Gai, and/or Gaq after dimerization [9][10] and induce accumulation of cAMP, activation of extracellular signal-regulated kinases (ERK) and protein kinase B (AKT) and calcium release [11]. PK2β, produced by alternative splicing of pk2 gene, is a biased ligand of prokineticin receptors: indeed, it binds preferentially PKR1 inducing Gas and Gaq and not Gai coupling [12][13].
PKRs are widely distributed throughout the body: PKR1 is mainly expressed in peripheral tissues, including endocrine glands and organs of the reproductive system, spleen, gastrointestinal tract, heart, lungs, and cells of the immune system (such as neutrophils and macrophages). In the CNS, while PKR2 is widely expressed [14], PKR1 is present only in some areas of the brain [15]. PKRs are also expressed on endothelial cells, where they modulate neovascularization (PKR1) and fenestration (PKR2) in adult animals, promoting angiogenesis and vascular permeability [16]. The prokineticin system is involved in a wide range of biological functions: neurogenesis [14], circadian rhythms [17], survival of cardiomyocytes [18], hematopoiesis, and regulation of the immune response [18]. In addition, prokineticins and their receptors are associated with diseases of various tissues: colon, testicular and prostate cancers, polycystic ovary syndrome (PCOS) [19][20][21] and congenital diseases (Kallmann syndrome and Hirschsprung disease) [22][23].
Based on the large amount of data collected over the last twenty years, it is possible to classify prokineticins as chemokines. Like chemokines, prokineticins are small peptides weighing 8–10 KDa that are basic, contain cysteine residues and act as potent chemotactic factors [18]. PK2 released in inflamed peritoneal tissue triggers macrophage recruitment, production of pro-inflammatory cytokines and reduction of anti-inflammatory cytokines through PKR1 activation [24].
PK2 also induces astrocyte migration, which is associated with a gradual shift in astrocytic phenotype A2 [25]. PK2 has been shown to regulate the signaling mechanism between neurons and astrocytes by inducing the phosphorylation of Signal Transducer and Activator of Transcription 3 (STAT3) after subarachnoid hemorrhage, thus acting as an endogenous mechanism for self-repair [26]. Prokineticin receptors are expressed in many tissues and are able to couple different G proteins inducing an extreme variability of responses. For this reason, dysregulation in the prokineticin system can lead to inflammatory and neuroinflammatory conditions, as reported in several studies.
2. Versatile Role of Prokineticins and Prokineticin Receptors in Neuroinflammation
Inflammatory pain is characterized by the release of various inflammatory mediators, such as cytokines and chemokines, from damaged or inflamed tissues and from nociceptive neurons themselves, which induce hypersensitivity both at the site of damage and in adjacent tissues [27].
In the animal model of inflammatory pain induced by the administration of Complete Freund Adjuvant (CFA) in the mouse paw, it has been shown that the development and duration of inflammatory pain correlates temporally with PK2 expression levels in inflamed tissues and that neutrophilic granulocytes are the major source of PK2 [14][28]. Granulocyte colony-stimulating factor (G-CSF), via activation of STAT3, is primarily responsible for PK2 overexpression in neutrophils invading inflamed tissue [29]. PK2 released in inflamed tissue triggers an innate and adaptive immune response. Innate response induces the recruitment of macrophages, the production of pro-inflammatory cytokines (IL-1β and IL-12) and reduction of anti-inflammatory cytokines (IL-10) [14][28]. Adaptative response modulates the cytokine production in T cells which assume a proinflammatory Th1 phenotype [14][28]. Secreted PK2 also regulates angiogenesis and vascular permeability [16][18][19] and induces intracellular Ca2+ mobilization, PKCɛ translocation and activation of TRPV1 [14][28], TRPA1 [30], sensitization of P2X channels [31] and suppression of GABAa-activated currents [32].
At the central level, the prokineticin system modulates pain by increasing GABA release in the periaqueductal gray (PAG), thereby increasing the firing activity of On cells and decreasing the firing activity of Off cells in the rostral ventral medulla (RVM) [14][28]. Moreover, PK2 worsens pain perception by suppressing the release of encephalins from neurons in the area postrema (AP), a circumventricular organ rich in PKR2 and lacking BBB [33].
Neuropathic pain is a type of chronic disabling pain due to damage or dysfunction of the peripheral or central nervous system. It is characterized by neuronal oversensitization and abnormal pain perception (allodynia and hyperalgesia). Treatment is currently difficult because the underlying mechanisms are not fully understood, although the involvement of proinflammatory cytokines and chemokines is becoming more apparent.
In animal models of neuropathic pain induced by sciatic nerve injury [spared nerve injury (SNI) and chronic constriction injury (CCI)], damage to the nerve results in tactile allodynia and thermal hyperalgesia, with upregulation of the PK2 and PKR2 receptors in the main stations involved in pain transmission: sciatic nerve, dorsal root ganglia (DRG) and dorsal horns of the spinal cord [14][28]. PK2, which is normally absent in the sciatic nerve, begins to be expressed as early as 3 days after injury in activated Schwann cells and infiltrating macrophages. Subsequently this overexpression spreads towards the DRG and spinal cord at an interval of 10 days after injury. In DRGs, PK2 is detectable in neurons and satellite cells, whereas in the spinal cord it is observed in activated astrocytes [14][28]. PK2 is stored in cytoplasmic vesicles and once released into the synapse by exocytosis, it activates PKR2 receptors at the postsynaptic level [14][28].
After peripheral nerve injury, PKR2 is also overexpressed in the sciatic nerve, DRG, and spinal cord, whereas PKR1 has overexpression restricted to the nerve [14][28]. The neuropathy induced by peripheral sciatic nerve damage leads to an increase in the permeability of both the “blood–nerve barrier” (BNB) and the “blood–spinal cord barrier” (BSCB) within 24–48 h [34]. The PKRs selective antagonist PC1 is also able to reduce the BSCB permeability; indeed as showed by the Blue Evans assay (Figure 1), the exudation induced by SNI is significantly reduced in the spinal cord after only 2 days of subcutaneous treatment with PC1 [35].

Figure 1. Increased blood–spinal cord barrier permeability (BSCB) after spinal nerve ligation (SNI) in mice. Lumbar spinal cord images of healthy mice compared to injured mice treated with saline or with PC1 (150 µg/kg sc, twice a day). In this Blue Evans assay exudation is evident in the spinal cord of SNI mice but PC1 is able to reduce it already after two days of treatment.
Painful neuropathy is a critical side effect of many chemotherapeutic agents. Mice injected intraperitoneally with bortezomib [36] or vincristine [37] develop allodynia and hyperalgesia, which correlates with strong activation of the prokineticin system activation of macrophage and glial markers, and sustained overproduction of cytokines in the sciatic nerve, DRG, and spinal cord. Moreover, in vitro in DRG neurons, the use of the PKRs antagonist PC1 was shown to prevent the neurotoxic effects of chemotherapeutic agents affecting the reduction in total neurite length induced by bortezomib and vincristine [38]. In vivo, subchronic administration of PC1 in mice exposed to all different models of neuropathy reduces both thermal hyperalgesia and mechanical and tactile allodynia by reducing PK2 overexpression in the different pain stations, restoring physiological levels of pro- and anti-inflammatory cytokines in both the periphery and spinal cord, and reducing spinal glial activation [35][36].
These results indicate that the prokineticin system represents a new potential therapeutic target to combat peripheral neuropathy.
3. Conclusions
Neuroinflammation is defined as an inflammatory response within the brain or the spinal cord associated with activation of the glia with significant production of cytokines and chemokines, infiltration of peripheral immune cells, edema, increased permeability and disruption of the blood–brain barrier. This damage can lead to infiltration of immune cells into the brain with consequent exacerbation of central inflammation.
Neuroinflammation, which is seen in obesity, diabetes, Parkinson’s and Alzheimer’s, causes several overlapping neurodegenerative mechanisms, including oxidative stress, mitochondrial dysfunction, and inflammation [39][40].
The prokineticin system is a central players in neuroinflammation (Figure 2) and could be a novel therapeutic target for neuroinflammatory diseases. However, since the system is present in different tissues and is involved in numerous physiological processes, the use of a drug capable of antagonizing the pro-inflammatory effect of prokineticins could also induce the onset of side effects.
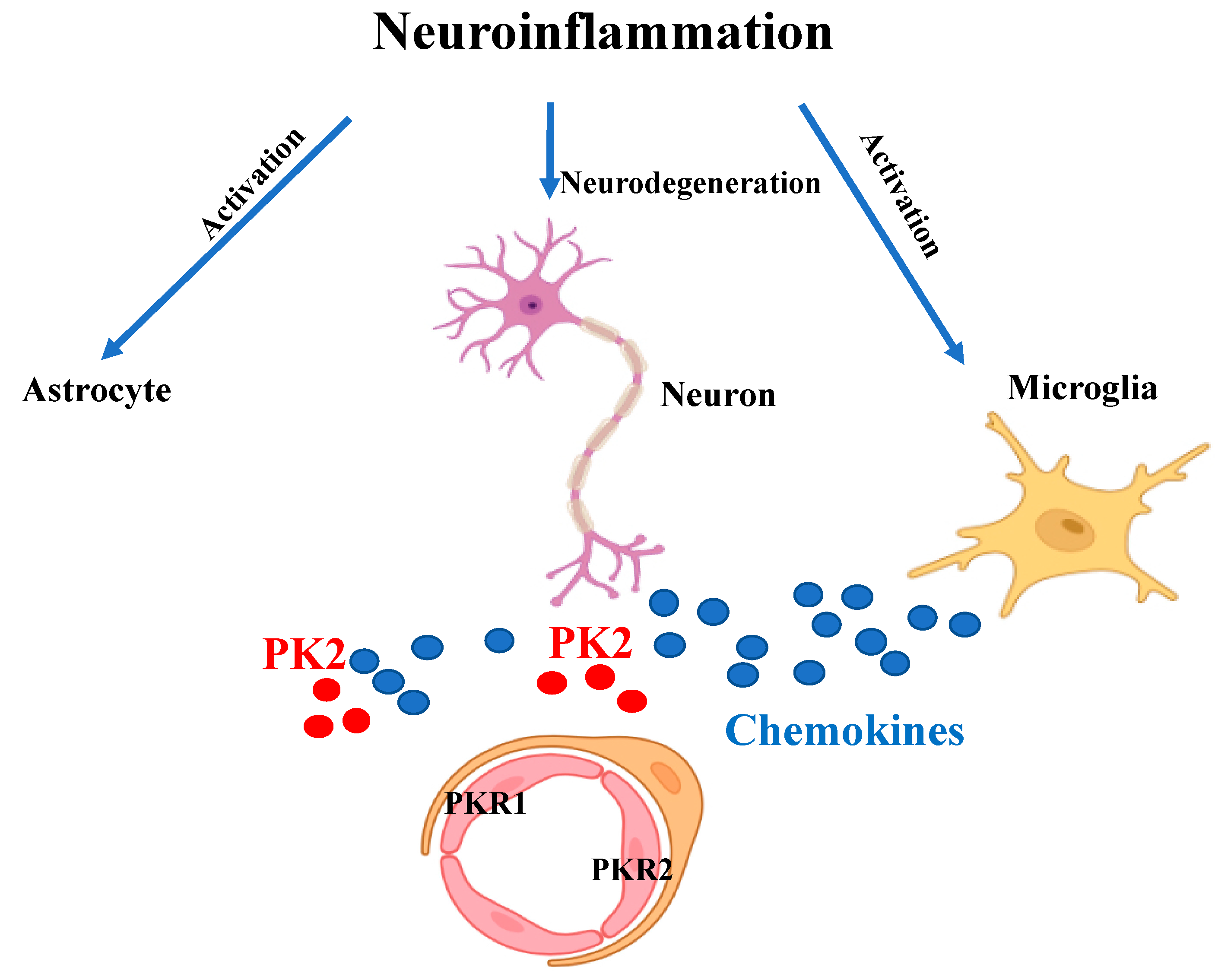
Figure 2. Schematic representation of the inflammatory state. Neuroinflammation leads to neurodegeneration and to over-activation of microglia and astrocytes which release pro-inflammatory cytokines and chemokines (blue circles). The chemokine-like protein PK2 (red circles) is secreted by neurons and astrocytes. Upon binding to PKR1 and PKR2 receptors on endothelial cells it promotes angiogenesis and vascular permeability.
References
- Gozales, H.; Pacheco, R. T-cell-mediated regulation of neuroinflammation involved in neurodegenerative diseases. J. Neuroinfl. 2014, 11, 201.
- Bagyinszkya, E.; Van Giaua, V.; Shima, K.; Sukb, K.; Ana, S.S.A.; Kim, S.Y. Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J. Neurol. Sci. 2017, 376, 242–254.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–167.
- Sofroniew, M.V. Astrocyte barriers to neurotoxic inflammation. Nat. Rev. Neurosci. 2015, 16, 249–263.
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967.
- Kaser, A.; Winklmayr, M.; Lepperdinger, G.; Kreil, G. The AVIT protein family. Secreted cysteine-rich vertebrate proteins with diverse functions. EMBO Rep. 2003, 4, 469–473.
- Miele, R.; Lattanzi, R.; Bonaccorsi di Patti, M.C.; Paiardini, A.; Negri, L.; Barra, D. Expression of Bv8 in Pichia pastoris to identify structural features for receptor binding. Protein Expr. Purif. 2010, 73, 10–14.
- Lattanzi, R.; Sacerdote, P.; Franchi, S.; Canestrelli, M.; Miele, R.; Barra, D.; Visentin, S.; DeNuccio, C.; Porreca, F.; De Felice, M.; et al. Pharmacological activity of a Bv8 analogue modified in position 24. Br. J. Pharmacol. 2012, 166, 950–963.
- Marsango, S.; Bonaccorsi di Patti, M.C.; Barra, D.; Miele, R. Evidence that prokineticin receptor 2 exists as a dimer in vivo. Cell. Mol. Life Sci. 2011, 68, 2919–2929.
- Sposini, S.; Caltabiano, G.; Hanyaloglu, A.C.; Miele, R. Identification of transmembrane domains that regulate spatial arrangements and activity of prokineticin receptor 2 dimers. Mol. Cell. Endocrinol. 2015, 399, 362–372.
- Désaubry, L.; Kanthasamy, A.G.; Nebigil, C.G. Prokineticin signaling in heart-brain developmental axis: Therapeutic options for heart and brain injuries. Pharm. Res. 2020, 160, 105190.
- Chen, J.; Kuei, C.; Sutton, S.; Wilson, S.; Yu, J.; Kamme, F.; Mazur, C.; Lovenberg, T.; Liu, C. Identification and pharmacological characterization of prokineticin 2 beta as a selective ligand for prokineticin receptor 1. Mol. Pharmacol. 2005, 67, 2070–2076.
- Lattanzi, R.; Maftei, D.; Negri, L.; Fusco, I.; Miele, R. PK2β ligand, a splice variant of prokineticin 2, is able to modulate and drive signaling through PKR1 receptor. Neuropeptides 2018, 71, 32–42.
- Negri, L.; Ferrara, N. The prokineticins: Neuromodulators and mediators of inflammation and myeloid cell-dependent angiogenesis. Physiol. Rev. 2018, 98, 1055–1082.
- Cheng, M.Y.; Leslie, F.M.; Zhou, Q.Y. Expression of prokineticins and their receptors in the adult mouse brain. J. Comp. Neurol. 2006, 498, 796–809.
- Nebigil, C.G. Prokineticin receptors in cardiovascular function: Foe or friend. Trends Cardiovasc. Med. 2009, 19, 55–60.
- Cheng, M.Y.; Bullock, C.M.; Li, C.; Lee, A.G.; Bermak, J.C.; Belluzzi, J.; Weaver, D.R.; Leslie, F.M.; Zhou, Q.Y. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachi- asmatic nucleus. Nature 2006, 417, 405–410.
- LeCouter, J.; Zlot, C.; Tejada, M.; Peale, F.; Ferrara, N. Bv8 and endocrine gland-derived vascular endothelial growth factor stimulate hematopoiesis and hematopoietic cell mobilization. Proc. Natl. Acad. Sci. USA 2004, 101, 16813–16818.
- Monnier, J.; Samson, M. Prokineticins in angiogenesis and cancer. Cancer Lett. 2010, 296, 144–149, Epub 14 July 2010.
- Meng, L.; Yang, H.; Jin, C.; Quan, S. miR-28–5p suppresses cell proliferation and weakens the progression of polycystic ovary syndrome by targeting prokineticin-1. Mol. Med. Rep. 2019, 20, 2468–2475, Epub 1 July 2019.
- Alfaidy, N.; Brouillet, S.; Rajaraman, G.; Kalionis, B.; Hoffmann, P.; Barjat, Y.; Benharouga, M.; Murthi, P. The Emerging Role of the Prokineticins and Homeobox Genes in the Vascularization of the Placenta: Physiological and Pathological Aspects. Front. Physiol. 2020, 12, 591850.
- Dodé, C.; Rondard, P. PROK2/PROKR2 Signaling and Kallmann Syndrome. Front. Endocrinol. 2013, 4, 19.
- Ruiz-Ferrer, M.; Torroglosa, A.; Núñez-Torres, R.; De Agustín, J.C.; Antiñolo, G.; Borrego, S. Expression of PROKR1 and PROKR2 in Human Enteric Neural Precursor Cells and Identification of Sequence Variants Suggest a Role in HSCR. PLoS ONE 2011, 6, e23475.
- Martucci, C.; Franchi, S.; Giannini, E.; Tian, H.; Melchiorri, P.; Negri, L.; Sacerdote, P. Bv8, the amphibian homologue of the mammalian prokineticins, induces a proinflammatory phenotype of mouse macrophages. Br. J. Pharmacol. 2006, 147, 225–234.
- Neal, M.; Luo, D.; Harischandra, D.S.; Gordon, R.; Sarkar, S.; Jin, H.; Anantharam, V.; Désaubry, L.; Kanthasamy, A.; Kanthasamy, A. Prokineticin-2 promotes chemotaxis and alternative A2 reactivity of astrocytes. Glia 2018, 66, 2137–2157.
- Ma, M.; Li, H.; Wu, J.; Zhang, Y.; Shen, H.; Li, X.; Wang, Z.; Chen, G. Roles of Prokineticin 2 in Subarachnoid Hemorrhage-Induced Early Brain Injury via Regulation of Phenotype Polarization in Astrocytes. Mol. Neurobiol. 2020, 57, 3744–3758.
- Julius, D.; Basbaum, A.I. Molecular mechanisms of nociception. Nature 2001, 413, 203–210.
- Franchi, S.; Sacerdote, P.; Panerai, P. The prokineticin system: An interface between neural inflammation and pain. Neurol. Sci. 2017, 38, S27–S30.
- Shojaei, F.; Ferrara, N. Role of the microenvironment in tumor growth and in refrac- toriness/resistance to anti-angiogenic therapies. Drug Resist. Updat. 2008, 11, 219–230.
- Maftei, D.; Vellani, V.; Artico, M.; Giacomoni, C.; Severini, C.; Lattanzi, R. Abnormal Pain Sensation in Mice Lacking the Prokineticin Receptor PKR2: Interaction of PKR2 with Transient Receptor Potential TRPV1 and TRPA1. J. Neurosci. 2020, 427, 16–28.
- Ren, C.; Qiu, C.Y.; Gan, X.; Liu, T.T.; Qu, Z.W.; Rao, Z.; Hu, W.P. Prokineticin 2 facilitates mechanical allodynia induced by α,β-methylene ATP in rats. Eur. J. Pharmacol. 2015, 767, 24–29.
- Xiong, Y.C.; Li, X.M.; Wang, X.J.; Liu, Y.Q.; Qiu, F.; Wu, D.; Gan, Y.B.; Wang, B.H.; Hu, W.P. Prokineticin 2 suppresses GABA-activated current in rat primary sensory neurons. Neuropharmacology 2010, 59, 589–594.
- Ingves, M.V.; Ferguson, A.V. Prokineticin 2 modulates the excitability of area postrema neurons in vitro in the rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R617–R626.
- Montague-Cardoso, K.; Malcangio, M. Changes in blood–spinal cord barrier permeability and neuroimmune interactions in the underlying mechanisms of chronic pain. Pain Rep. 2021, 6, e879, PMCID:PMC8108584.
- Congiu, C.; Onnis, V.; Deplano, A.; Salvadori, S.; Marconi, V.; Maftei, D.; Negri, L.; Lattanzi, R.; Balboni, G. A new convenient synthetic method and preliminary pharmacological characterization of triazinediones as prokineticin receptor antagonists. Eur. J. Med. Chem. 2014, 81, 334–340.
- Moschetti, G.; Amodeo, G.; Maftei, D.; Lattanzi, R.; Procacci, P.; Sartori, P.; Balboni, G.; Onnis, V.; Conte, V.; Panerai, A.; et al. Targeting prokineticin system counteracts hypersensitivity, neuroinflammation, and tissue damage in a mouse model of bortezomib-induced peripheral neuropathy. Neuroinflammation 2019, 16, 89.
- Moschetti, G.; Amodeo, G.; Paladini, M.S.; Molteni, R.; Balboni, G.; Panerai, A.; Sacerdote, P.; Franchi, S. Prokineticin 2 promotes and sustains neuroinflammation in vincristine treated mice: Focus on pain and emotional like behavior. Brain Behaviur Immun. 2019, 82, 422–431.
- Moschetti, G.; Kalpachidou, T.; Amodeo, G.; Lattanzi, R.; Sacerdote, P.; Kress, M.; Franchi, S. Prokineticin Receptor Inhibition with PC1 protects Mouse Primary Sensory Neurons FromNeurotoxic Effects of Chemotherapeutic Drugs in vitro. Front. Immunol. 2020, 11, 2119.
- Pugazhenthi, S.; Limei, Q.L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 517–552.
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139, 136–153.
More
Information
Subjects:
Cell Biology; Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revision:
1 time
(View History)
Update Date:
18 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No