Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mayara Jorgens Prado | + 4567 word(s) | 4567 | 2021-12-29 07:12:34 | | | |
| 2 | Vivi Li | + 9 word(s) | 4576 | 2022-02-17 03:01:16 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jorgens Prado, M.; Pandey, A.; Ligabue-Braun, R.; Valandro Meneghetti, B. CYP21A2 Deficiency. Encyclopedia. Available online: https://encyclopedia.pub/entry/19554 (accessed on 26 July 2026).
Jorgens Prado M, Pandey A, Ligabue-Braun R, Valandro Meneghetti B. CYP21A2 Deficiency. Encyclopedia. Available at: https://encyclopedia.pub/entry/19554. Accessed July 26, 2026.
Jorgens Prado, Mayara, Amit Pandey, Rodrigo Ligabue-Braun, Bruna Valandro Meneghetti. "CYP21A2 Deficiency" Encyclopedia, https://encyclopedia.pub/entry/19554 (accessed July 26, 2026).
Jorgens Prado, M., Pandey, A., Ligabue-Braun, R., & Valandro Meneghetti, B. (2022, February 17). CYP21A2 Deficiency. In Encyclopedia. https://encyclopedia.pub/entry/19554
Jorgens Prado, Mayara, et al. "CYP21A2 Deficiency." Encyclopedia. Web. 17 February, 2022.
Copy Citation
Deficiency of 21-hydroxylase enzyme (CYP21A2) represents 90% of cases in congenital adrenal hyperplasia (CAH), an autosomal recessive disease caused by defects in cortisol biosynthesis. Computational prediction and functional studies are often the only way to classify variants to understand the links to disease-causing effects.
21-hydroxylase deficiency
congenital adrenal hyperplasia
CYP21A2
functional characterization
1. Introduction
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disease caused by defects in steroid biosynthesis [1]. More than 90% of reported CAH cases are due to 21-hydroxylase (CYP21A2) deficiency (OMIM # 201910). The CYP21A2 is a member of the cytochrome P450 superfamily and has 495 amino acids forming thirteen α-helix (A-M) and nine β-sheets [2][3]. This protein is located in the endoplasmic reticulum of the adrenal cortex and has a role in both the glucocorticoid and mineralocorticoid biosynthesis by the hydroxylations of 17-hydroxyprogesterone into 11-deoxycortisol and progesterone into 11-deoxycorticosterone, which are then converted into cortisol and aldosterone (Figure 1) [1]. Therefore, defects in CYP21A2 affect both the mineralocorticoid and glucocorticoid biosynthesis, besides the increase in sex steroids biosynthesis due to changes in the steroidogenesis pathway by elevated levels of sex steroid precursors [4][5].
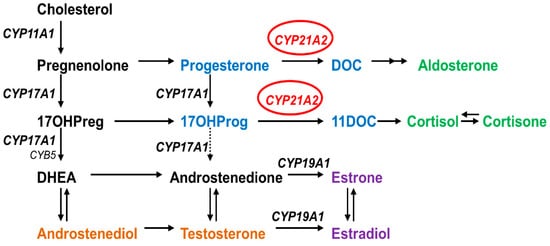
Figure 1. Role of CYP21A2 in human steroid biosynthesis. CYP21A2 is a heme containing cytochrome P450 protein that requires cytochrome P450 reductase as a redox partner for its metabolic reactions [6]. Together with CYP11A1 in mitochondria and CYP17A1 and CYP19A1 in the endoplasmic reticulum, CYP21A2 directs the biosynthesis of steroids.
Glucocorticoid and mineralocorticoid levels determine the CYP21A2 deficiency phenotype, which can be (1) salt wasting, when both types of steroids are not produced, causing the adrenal crisis by electrolytic deregulation with infant mortality risks and severe virilization by elevated sex steroids; (2) simple virilizing, when there is some residual synthesis of both glucocorticoid and mineralocorticoid that is enough to prevent the adrenal crisis; or (3) non-classical (mild form) when just the glucocorticoid synthesis is partially affected and is linked to hyperandrogenism and mild late-onset CAH. The first two are the classical forms of CAH, which have a worldwide incidence ranging from 1:14,000 to 1:18,000 live births [7]. The non-classic form has a frequency around 1:100 to 1:1000 [8]. The frequencies of CAH vary with ethnicity and geographic population groups. However, the description of SW vs. SV, SV vs. NC, etc. is rather arbitrary, hence caution must be employed when following [9]. The diagnosis of CYP21A2 deficiency is confirmed by steroid profile, mainly 17OHP in the first screening, which becomes elevated [1][7]. However, 17OHP can be altered by a deficiency in other enzymes in the steroidogenic pathway, in premature infants, and in unrelated diseases causing physiologic stress [7][8][10][11][12][13][14][15]. Therefore, the molecular diagnosis is essential for the confirmation of complex cases and follow-up management of asymptomatic CAH, avoiding unnecessary treatment, along with genetic counseling [11][16][17].
The CYP21A2 gene is located on the short arm of chromosome 6 (6p21.3), situated 30 kb apart from its pseudogene (CYP21A1P). These genes share 98% sequence identity for the exons and 96% among the introns [18]. Besides that, 95% of pathogenic variants of CYP21A2 originate in recombination events [3]. More than 1300 variants in the CYP21A2 have been reported in the human gene mutation database (HGMD), and more than 200 of these are described to affect human health. With high variability between different ethnic groups and single nucleotide variants (SNVs), missense and nonsense account for half of the total variations [19][20].
CYP21A2 variants are classified according to the impact on the enzyme activity. Group Null consists of deletions or nonsense variants that critically affect the enzyme activity, resulting in a complete loss of function due to altered enzyme stability, steroid or heme binding, and membrane anchoring. The most common variants are thirty kilobase deletion (30-kb del), eight base pairs, Cluster E6 (p.I237N, p.V238E, p.M240K), p.Q319X, p.R357W, and p.L307fs [3]. Group A is composed of a variant in which the enzyme activity is minimal, around 0–1%. This group is represented by an intron variant IVS2-13A/C>G, which is created by an additional splice acceptor site causing retention of 19 intronic nucleotides of the intron 2 [21][22]. Homozygous or compound heterozygote variants with the null group are often associated with the salt-wasting form, but approximately 20% of cases have a simple virilizing phenotype [3]. Group B has a residual activity of 1–10%, which is enough to prevent adrenal crisis. The p.I173N variant is representative of this group and is associated with the simple virilizing form of 21-hydroxylase deficiency [23]. Finally, group C has an enzyme activity of about 20-60% and is associated with the mild form of CAH. The variants common in this group are p.V282L, p.P454S, and p.P31L, which show phenotype variability [24].
The biochemical correlation for CYP21A2 activity works well for the CAH diagnosis. However, external factors or a combination of genetic diseases can change the steroid levels, making genotype elucidation an important tool for patient management [11][17]. In general, there is a good genotype-phenotype correlation for CAH, and the elucidation of new variants found in each population is important to improve the correct treatment [25][26]. Initially, the bovine CYP21A2 structure was used as a template to elucidate the impact of variants damage from 2011 to 2015 (PDB ID 3QZ1), but the human CYP21A2 crystal structures have recently become available. Human CYP21A2 is deposited in the RSCB Protein Data Bank (PDB) under entries 4Y8W and 5VBU, with two different steroid ligands (progesterone in 4Y8W and 17-hydroxyprogesterone in 5VBU). The human structure has just one steroid-binding site, which is different from the bovine enzyme, which has two catalytic sites [2][27].
2. Variant Collection and Description
Researchers selected six missense variants that had patient genotype and phenotype available in researchers' literature review after the initial screening described in methods. Four of these variants were found in Brazilian (p.P35L, p.L199P, p.E352V, and p.R484L) and two in Portuguese (p.W202R and p.P433L) populations. Genetic and clinical data describing the carriers of the selected SNVs are summarized in Table 1. These data were collected from the original papers.
Table 1. Genetic and clinical features of the subjects carrying the selected SNVs (bold) in the CYP21A2 gene. All data are from the original papers. Normal values for 17α-hydroxyprogesterone (17OHP) are <30 nmol/L, serum sodium between 132–142 mmol/L and potassium 3.6–6.1 mmol/L. LGC, large gene conversion; 30-kb del, large deletion from CYP21A1P to CYP21A2 gene; nd, non-determined; DSD, ambiguous/atypical genitalia; AC, adrenal crisis; M, male; F, female. a Age at diagnosis, b non-diluted measurements (reference value 5.97 nmol/L), c after ACTH stimulation.
| Genotypes | Phenotype | Clinical Data | Sex | 17OHP (nmol/L) | Na+/K+ (mmol/L) | Age a | Reference | |
|---|---|---|---|---|---|---|---|---|
| Allele 1 | Allele 2 | |||||||
| P35L, H63L, 30-kb del | LGC | SW | vomiting, dehydration | M | 49.3 | 116/9.2 | 2.8 m | [28] Coeli et al. (2010) |
| P35L, H63L, 30-kb del | Q319X | SW | vomiting, dehydration | M | >25 b | 120/6.8 | 13 m | [28] Coeli et al. (2010) |
| P35L, H63L, 30-kb del | c.920_921insT | SW | DSD (Prader IV) | F | >6 b | 119/5.3 | 15 d | [28] Coeli et al. (2010) |
| P35L, H63L, 30-kb del | R357W | SW | vomiting, dehydration, AC | M | >200 b | 119/9.7 | 28 d | [28] Coeli et al. (2010) |
| L199P | normal | ND | DSD (Prader I) | F | 408.5 | nd | [29] Silveira et al. (2009) | |
| W202R | LGC | SW | DSD, salt wasting | F | nd | nd | <2 m | [30] Santos-Silva et al. (2019) |
| E352V | E6 cluster | SV | nd | F | nd | nd | nd | [16] De Carvalho et al. (2016) |
| E352V | IVS-13A/C>G | SW | nd | M | nd | nd | nd | [16] De Carvalho et al. (2016) |
| E352V | IVS-13A/C>G | SW | nd | M | nd | nd | nd | [16] De Carvalho et al. (2016) |
| E352V | G425S | SV | nd | M | nd | nd | nd | [16] De Carvalho et al. (2016) |
| P433L | P454S | NC | nd | F | 42.7 c | nd | 17 y | [31] Carvalho et al. (2012) |
| R484L, IVS-13A/C > G | Del CYP21A2 | SW | DSD (Prader III-IV) | F | 1537.2 | 134/4.9 | <4 m | [29] Silveira et al. (2009) |
| R484L, IVS-13A/C > G | Q319X | SW | undefined | M | 1358.7 | 130/6.2 | <9 m | [29] Silveira et al. (2009) |
The variant p.P35L was identified in four Brazilian patients as compound heterozygous with other mutations (Table 1). One allele presents a mutation with symptoms of severe enzyme damage, while the other alleles carried three mutations: p.P35L, p.H63L, and 30-kb del. It was concluded that the p.P35L origin was not from the same genetic event of the p.H63L and 30-kb del, as it was not present in the pseudogene screening. Therefore, researchers analyzed this variant alone.
The variant p.L199P was found as heterozygous in a female patient with mild clitoromegaly atypical genitalia and elevated 17OHP levels detected in the Brazilian newborn screening program (Table 1). According to the authors, the child remained asymptomatic at 3.3 years old, and the clitoromegaly and the high 17OHP levels detected in the screening were likely due to the premature birth. The impact of this new variant has been unknown, as it was not possible to infer the consequence once it was identified in heterozygosis.
The variant p.W202R was found as compound heterozygous with large gene conversion in a Portuguese female (Table 1). This patient presented atypical genitalia and salt wasting, being diagnosed with classical CAH at <2 months old.
The variant p.E352V was identified in a compound heterozygous state in four classical CAH patients from Brazil (Table 1), two of them with the same intronic mutation, IVS2-13A/C>G heterozygous with p.E52V. These two patients showed the classical CAH form with salt-wasting. One female presented p.R484L as compound heterozygous with Cluster E6. These patients presented the simple virilizing CAH form. The latter has the p.R484L and p.G425S, which resulted in the simple virilization form of CAH.
The variant p.P433L was identified in a Portuguese female who presented it as compound heterozygous with the mild mutation p.P454S (Table 1). This patient was diagnosed at 17 years old with a nonclassical form of CAH after ACTH stimulation, which indicated the 17OHP elevation characteristic of that form of CAH.
The variant p.R484L was found as compound heterozygous in two Brazilian patients (Table 1). Both present classical CAH form, with salt-wasting and high 17OHP levels detected during newborn screening. One allele in both cases has p.R484L together with a splice variant IVS2-13A/C>G. The second allele in one patient was a CYP21A2 deletion and in the other p.Q319X.
3. Computational Characterization Indicated the Structural Impact of the SNVs
Researchers performed a screening with five predictive tools that have different approaches, PolyPhen-2, SNAP2, MutPred2, Meta-SNP, and PredictSNP. Results obtained for the six variants chosen in this work are shown in Table 2. The variants p.L199P, p.E352V, and p.R484L were predicted to damage the protein by all predictor tools, while p.P35L had damage predicted by four tools, p.W202R by three, and p.P433L by two.
Table 2. Prediction of the possible impact on the structure and/or function of CYP21A2 by genetic variants. Seven variants were used to validate the method, with four polymorphisms (●) and three with a known negative impact (↓). a SNV nomenclature according to UniProt ID Q16874-1. Scores ≥0.5 by PolyPhen-2, MutPred2, and Meta-SNP indicate protein damage. SNAP2 scores >50 indicate a strong signal for effect, between 50 and −50 weak signal and <−50 strong signal for neutral effect.
| SNV a | In Silico Tools | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| dbSNP rs# | PolyPhen-2 | Score | SNAP2 | Score | MutPred2 | Score | Meta-SNP | Score | PredictSNP | Enzyme Activity % of Control |
|
| ● R103L | rs6474 | Benign | 0.023 | Neutral | −47 | Neutral | 0.191 | Neutral | 0.310 | Neutral | 120 |
| ● D184E | rs397515531 | Benign | 0.000 | Neutral | −85 | Neutral | 0.260 | Neutral | 0.412 | Neutral | 100 |
| ● S269T | rs6472 | Benign | 0.016 | Neutral | −93 | Neutral | 0.076 | Neutral | 0.340 | Neutral | 103 |
| ↓ I173N | rs6475 | Damage | 1.000 | Effect | 70 | Pathogenic | 0.838 | Disease | 0.811 | Deleterious | 1.1 |
| ↓ V282L | rs6471 | Benign | 0.273 | Neutral | −76 | Neutral | 0.084 | Neutral | 0.344 | Neutral | 16.4 |
| ↓ R427H | rs151344504 | Damage | 1.000 | Effect | 82 | Pathogenic | 0.821 | Disease | 0.827 | Deleterious | 0.5 |
| P35L | rs200648381 | Damage | 0.988 | Effect | 13 | Pathogenic | 0.676 | Disease | 0.624 | Neutral | 13 |
| L199P | Damage | 0.997 | Effect | 68 | Pathogenic | 0.875 | Disease | 0.711 | Deleterious | 10.3 | |
| W202R | Damage | 1.000 | Effect | 45 | Pathogenic | 0.577 | Neutral | 0.451 | Neutral | 1.2 | |
| E352V | Damage | 0.693 | Effect | 90 | Pathogenic | 0.919 | Disease | 0.937 | Deleterious | 1.1 | |
| P433L | rs751456004 | Benign | 0.402 | Effect | 15 | Pathogenic | 0.614 | Neutral | 0.372 | Neutral | 7.5 |
| R484L | Damage | 1.000 | Effect | 81 | Pathogenic | 0.690 | Disease | 0.748 | Deleterious | 3.4 | |
Among the variants with known activity, the neutral variants (p.R103K, p.D183E, and p.S269T) agreed with all tools. However, among the three variants known to cause damage, researchers had different results. The two severe variants (p.I173N and p.R427H) were predicted correctly by all tools; however, the mild (p.V282L) variant was wrongly classified by all of them. The second screening about the protein stability of all variants presented a variation of ∆G, from −1.47 kcal/mol (p.R484L) to 1.23 kcal/mol (p.E352V) (Table 2 and Table 3).
Table 3. Summary of structural and functional data obtained for the six variants studied shows that all of them can be related to CYP21A2deficiency. a SNV nomenclature according to UniProt ID Q16874-1. b Two hydrogen bonds with E355. H, hydrogen-bound.
| SNV a | Structure/Protein Localization | Physicochemical Properties | ΔΔG (kcal/mol) | Enzyme Activity | The Molecular Mechanism (Hypothesis) | Mutation Group | |
|---|---|---|---|---|---|---|---|
| WT | MUT | (Mean ± SD) | |||||
| P35L | N-term coil/Region of the membrane protein orientation | Polar residue with a ring at the N-term | Hydrophobic; Short side chain; Folding interactions | −0.58 | 13.2 ± 1.56% | Disturbs the protein orientation in the membrane | C |
| L199P | F-helix/Close to the active site; α-helix stabilization | Hydrophobic; Short side chain; Folding Interactions; H: I195 and S203 | Polar residue with a ring at the N-term; H: S203 | 0.79 | 10.3 ± 0.33% | Breaks the secondary structure close to the active site | C |
| W202R | Turn of the F-helix/close to the active site and steroid-bound residue (R234) | Hydrophobic; Large, rigid aromatic group; H: V198 | Hydrophobic; Long, flexible, and posit. charged side chain; Ionics-bound; H: V198 | 1.11 | 1.2 ± 0.34% | Positive charge addition causes disorder in heme binding | B |
| E352V | K-helix/ERR-triad associated with the heme group | Hydrophobic; Long, slightly flexible side chain; Strongly neg. charged; Ionics-bound; Fix metal iron; H: A348, R355 b, L356 and W406 | Hydrophobic; Short side chain; Folding interactions; H: A348 and L356 | 1.23 | 1.1 ± 0.11% | Disorder of the ERR-triad decreases heme-binding and stability | B |
| P433L | L-helix/Adjacent to essential residues for the heme bond | Polar residue with a ring at the N-term; H: A435 | Hydrophobic; Short side chain; Folding interactions; H: A435 | 0.39 | 7.5 ± 0.67% | Changes the natural orientation of heme-binding residues R427 and C429 | B |
| R484L | C-term coil/Hydrophobic cluster with ionic connection | Hydrophobic; Long, flexible, and posit. charged side chain; Ionics-bound; H: Q482, A449, and M486 | Hydrophobic; Short side chain; Folding interactions; H: A449 | −1.47 | 3.4 ± 0.8% | Loss of hydrophobic organization at the C-term region | B |
4. Amino Acid Chemical Proprieties, Conservation, and Structural Damage
Researchers analyzed the amino acid chemical properties to assess the characteristics of each exchange and the conservation of that residue across species to determine how it has been retained during the evolution of CYPs P450. Researchers worked with 200 CYP 450 sequences homologous to human CYP21A2. The structural damage was characterized by Gibbs’s free energy and gain/loss of hydrogen bonds.
Proline at amino acid position 35 is located in a coil on the protein surface (Figure 2 and Figure 3). This residue shows a high conservation score, with only two residues found on homologous sequences (proline and guanine) (Figure 4 and Table 4). The variant p.P35L has the amino acid properties changed from a polar and uncharged to a nonpolar and aliphatic residue, increasing the structural stability, ∆∆G −0.58 kcal/mol (Table 3). PolyPhen-2, SNAP, Meta-SNP, and MutPred2 predicted the effect of this mutation on the protein stability and functionality. Besides that, MutPred2 predicts two structural effects: gain of helix and alteration of transmembrane features. PredictSNP classified this variant as neutral (Table 2).
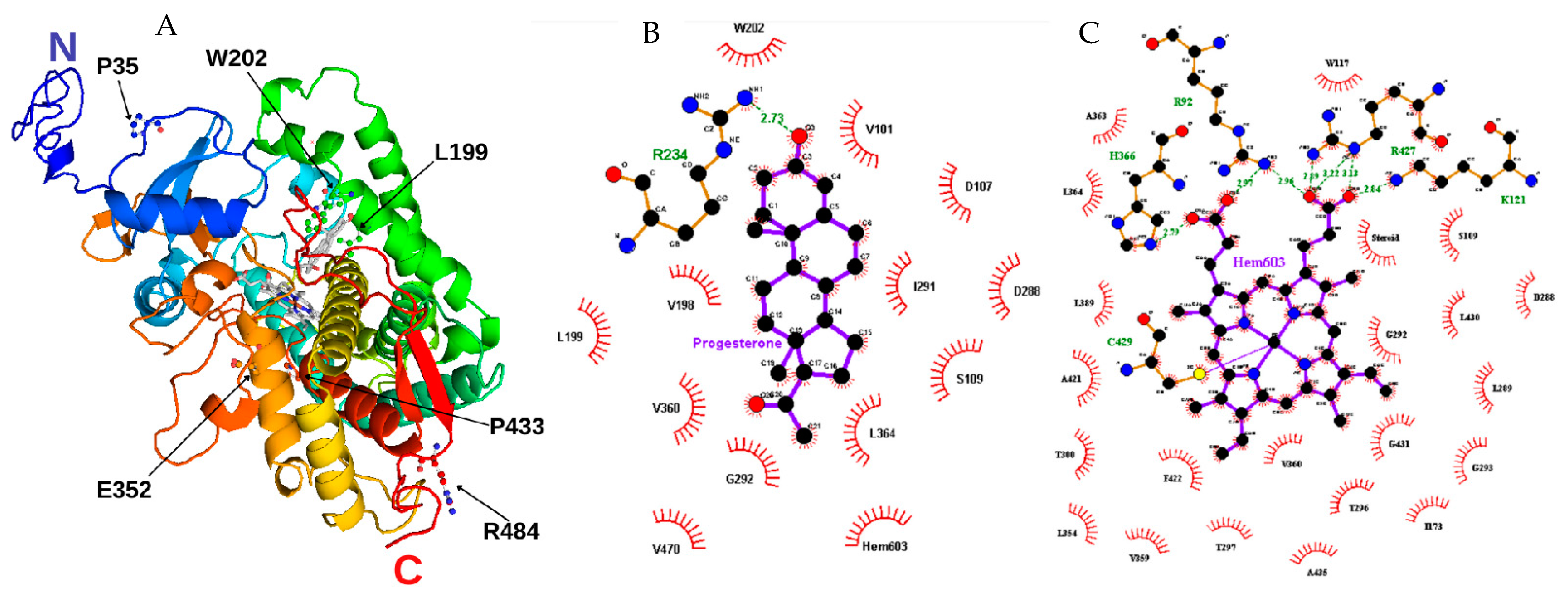
Figure 2. Structural model of CYP21A2. (A) Researchers built the missing extremity based on PDB ID 4Y8W with I-TASSER and visualized by PyMOL. The amino acids mutated in researchers' study are identified with black arrows. Progesterone and protoporphyrin containing Fe3+ are in the middle of the structure colored in grey. (B) Contact map with progesterone binding site in 21OH structure. The studied residue p.W202 is shown at the top. (C) Contact map with heme (protoporphyrin containing Fe) binding site in 21OH structure. The residues p.C429 and p.R427 are near the studied residue p.P433. Heme and progesterone contact maps were built with the structure of CYP21A2 (PDB # 4Y8W). Residue ligand contact maps were generated with LigPlot+ v.2.2.4 software using as maximum hydrogen -acceptor/-donor distance 2.7 Å and 3.35 Å (green line), respectively, while for non-bonded, the minimum contact distance was 2.9 Å and the maximum 3.9 Å.
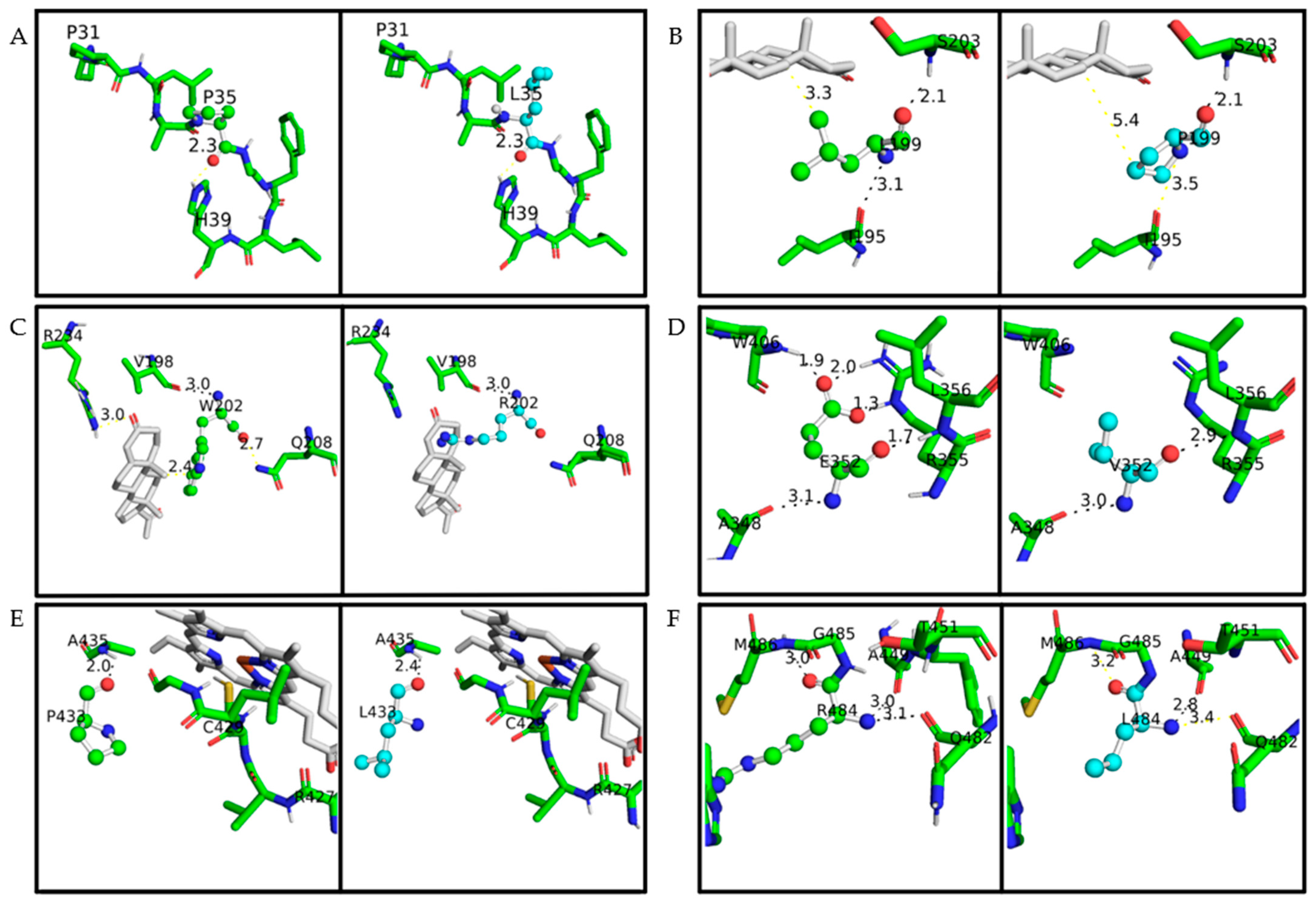
Figure 3. A closeup of the amino acid studied on the CYP21A2 structure. The protein structure is based on PDB ID 4Y8W. Wild-type amino acids are colored in green and mutated in cyan; in both of them, oxygen is red, nitrogen is blue, and sulfur is yellow. The distance measured between the main amino acids connections is with less than 4 Å. Hydrogen bonds are represented with black lines, while any other measurement between atoms is represented with yellow lines. Progesterone and heme are colored grey. (A) p.P35 and p.P35L, (B) p.L199 and p.L199P, (C) p.W202 and p.W202R, (D) p.E352 and p.E352V, (E) p.P433 and p.P433L, (F) p.R484 and p.R484L.
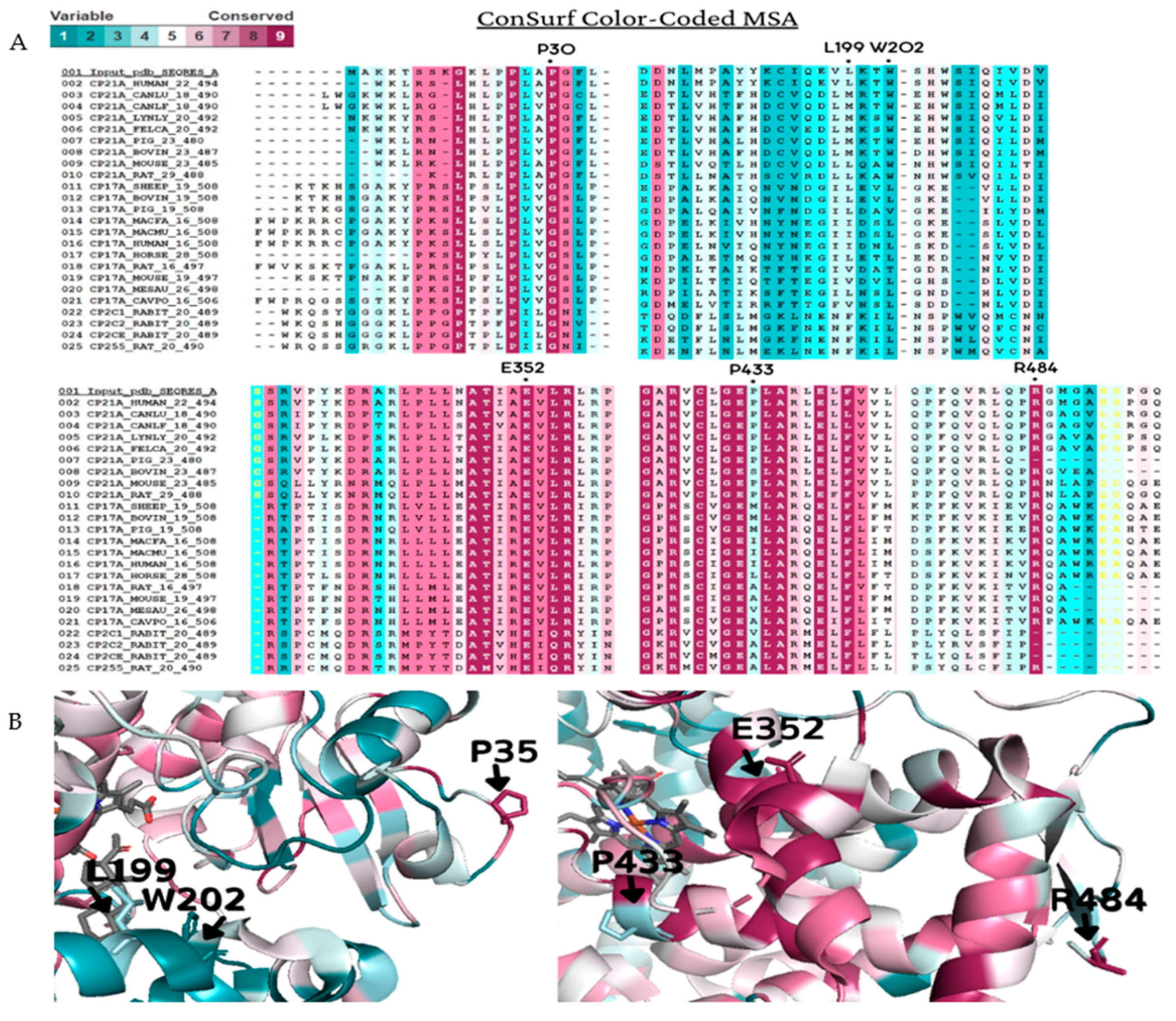
Figure 4. Amino acid conservation of the human CYP21A2. A CYP21A2 structure (PDB # 4Y8W) structure was used as a reference for ConSurf analysis. Multiple sequence alignment was built using CLUSTALW with 200-cytochrome P450 sequences homologous to the human protein 21-hydroxylase. The species considered for final alignment were from Homo sapiens, Bos taurus, Canis lupus familiaris, Cavia porcellus, Capra hircus, Felis catus, Gorilla gorilla gorilla, Lynx lynx, Mesocricetus auratus, Macaca fascicularis, Macaca mulatta, Mus musculus, Oryctolagus cuniculus, Ovis aries, Rattus norvegicus and Sus scrofa. (A) A representative part of the alignment from MSA. The specific positions of the six wild-type residues analyzed in this work are marked with a dot (●). (B) CYP21A2 structure with black marks on the six wild-types residues on the protein structure. Residues p.P35, p.E352, and p.R484 showed to be conserved, while p.L199, p.W202, and p.P433 are variable. Color-code: from pink (conserved) to cyan (variable).
Table 4. ConSurf amino acid conservation score. The score is from 1 (variable) to 9 (conserved). CI: Confidence interval of the score. a PDB ID 4Y8W. Details of any known variations in the amino acid positions studied are given in Table S3 (supplementary could be found in https://www.mdpi.com/1422-0067/23/1/296#supplementary).
| SNV a | Conservation Score | CI | Residue Variety |
|---|---|---|---|
| P35 | 9 | 9 to 8 | L, P, G |
| L199 | 3 | 4 to 2 | T, N, L, V, F, I, A, S, G, M |
| W202 | 1 | 2 to 1 | T, E, R, W, G, C, M, L, A, I, N, Q, D, V, F, S |
| E352 | 9 | 9 to 9 | E |
| P433 | 3 | 4 to 2 | R, K, G, M, L, V, H, A, S, I, T, P, E, Q |
| R484 | 9 | 9 to 9 | R, T, L |
Leucine at position 199 is localized at α-helix F, close to the CYP21A2 catalytic site <4 Å from heme, where it forms two hydrogen-bonds, one with p.S203 (on the loop between α-helix F and F’) and one with p.I195, on α-helix F (Figure 3 and Table S1). This residue shows mild conservation, being found in 10 different sequences at this position (Figure 4, Table 4). The variant p.L199P changes a nonpolar and aliphatic residue to a polar and uncharged residue, losing the hydrogen bond with p.I195 (Table S2). All predictor tools showed that this mutation causes damage to the protein, and MutPred2 predicted an alteration of coiled-coil and transmembrane regions; ∆∆G was 0.79 kcal/mol.
Tryptophan at position 202 is also located <4 Å from the CYP21A2 catalytic site, on the α-helix F with a hydrogen bond with p.V198 (Figure 3). This residue presented a low conservation score, being highly variable between its homologous sequences (Figure 4 and Table 4). The variant p.W202R changes a hydrophobic residue to a hydrophilic and positively charged residue, decreasing the CYP21A2 structural stability with a ∆∆G of 1.11 kcal/mol, but without losing the hydrogen bond (Table S2). Two predictor tools classified that residue replacement as neutral (Meta-SNP and PredictSNP), while the three others predicted damage to the protein. An altered coiled-coil region was predicted by MutPred2.
Glutamate at position 352 is located in α-helix K, presenting five hydrogen bonds, four on the same helix (one with p.A348 and p.L356 and two with p.R355) and one with a coil (p.W406) (Figure 3 and Table S1). This residue is highly conserved across species, and there is no other variation on CYP21A2 homologs (Figure 4 and Table 4). The variant p.E352V changes a negatively charged residue to a nonpolar aliphatic residue. The stability of the CYP21A2 structure was increased, and the changed residue lost three hydrogen bonds with W406 and p.R355 (Table S2). All the predictor tools showed that p.E352V results in protein damage (Table 2). The MutPred2 predicted alteration of an interface, loss of allosteric site at p.E352, and altered metal binding.
Proline at position 433 is located at α-helix M, where it has a hydrogen bond with p.A435 in the same helix (Figure 3 and Table S1). This residue is close to the central heme group, but it is not a conserved residue, as it is found interchangeable as 14 different residues among the CYP21A2 homologous group (Figure 4 and Table 4). However, this indicates the evolution of this residue across species with different roles and different redox partners. The exchange of a polar and uncharged amino acid with a nonpolar and aliphatic showed ∆∆G 0.39 kcal/mol (Table 3). The SNAP2 predictor tool showed damage in the protein with the variant and MutPred2 a gain of helix and loss of catalytic site at p.E432; however, the other three predictors indicated a neutral effect (Table 2).
Arginine at position 484 is located on the protein surface in a coil that makes a cluster with nine other amino acids. This residue has three hydrogen bonds, one with p.M486 in the same coil; one with p.Q482, located at stand β9; and one with p.A449, which makes the connection between α-helix M and β8 (Figure 3 and Table S1). This residue showed the highest conservation score, with just two other amino acids found in the homology analysis (Figure 4 and Table 4). The variant p.R484L changes a positively charged polar residue to a nonpolar and uncharged residue, losing the two hydrogen bonds with p.M486 and Q482 (Table S2). This amino acid exchange showed damage by all predictor tools used, and stability increased with a ∆∆G of -1.47 kcal/mol (Table 2 and Table 3). Loss of intrinsic disorder was predicted by MutPred2, as well.
5. Functional Testing for the Validation of In Silico Results
HEK293 cells were transfected with plasmids expressing CYP21A2 WT or variants p.P35L, p.L199P, p.W202R, p.E352V, p.P433L, p.R484L, p.I173N, and p.V282L as two controls with known activity (~2% and 18–60%, respectively) [4][23][24]. The CYP21A2 activity was quantified for both WT and variants. Using the TLC analysis, researchers could assess the conversion ratio of progesterone to 11-deoxycorticosterone and compare the activity of each variant with the WT enzyme (Figure 5A). Variants p.W202R and p.E352V showed only residual conversion (<2% enzyme activity), similar to p.I73N, which is associated with the classical form of CAH. Variants p.P35L and p.L199P showed partial activity, with the catalytic activities being 13.2% and 10.3% of the WT, similar to the p.V282L variant, which is associated with a nonclassical form of CAH. Variants p.P433L and p.R484L presented activity between the two controls, being 7.5% and 3.4% of the WT activity, respectively (Figure 5C).
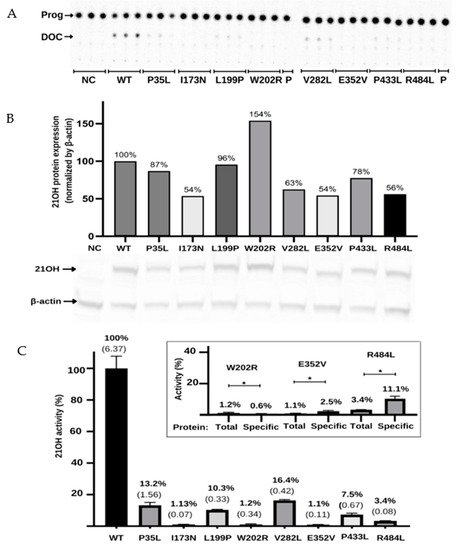
Figure 5. Functional characterization of the variants in the CYP21A2 gene by relative steroid conversion of progesterone (Prog; P) to 11-deoxyprogesterone (DOC). The activity was obtained using 21OH wild-type (WT) and mutated expressed in the HEK293t cell where researchers measured the steroid conversion by the percentage of radioactivity in DOC to the whole sample. (A) Thin-layer chromatography (TLC) shows that all variants reduce the activity of 21-hydroxylase (21OH). (B) Semiquantitative 21OH expression level obtained by western blot with anti-flag antibody for the 21OH (53 KDa) and normalized by β-actin (42 KDa) expression with anti-β-actin antibody. Negative control (NC) presents the basal HEK293 cells. (C) The 21OH activity expressed accordingly with TLC spots densitometry and related to the WT. Bars represent the standard error from three samples. Inside the box is plotted the activity (total) and the specific activity (specific) of the three variants that showed statistically significant difference (* p < 0.05) between these values. The specific activity was obtained by dividing the enzyme activity by the 21OH protein estimated by western blot. All results were analyzed on GraphPad software. * p value < 0.05.
To determine if the reduction in activity was associated with the decrease in protein expression, researchers quantified CYP21A2 protein in WT and variants using western blot assay (Figure 5B). Normalized by β-actin expression, researchers found the expression level was less than 50% of the WT for the p.P35L, p.V282L, p.E352V, and p.R484L variants, while p.W202R had 119% of WT. Therefore, researchers also calculated the specific activity of each variant compared to WT by dividing the activity obtained from the TLC results by the CYP21A2 expression level (Figure 5C). The specificity activity for p.W202R, p.E352V, and p.R484L present a significant difference (p < 0.05) when compared with the non-protein normalized activity. The variant p.W202R had a decrease in activity to <1%, while p.E352V and p.R484L had an increase in activity by two-fold and three-fold, respectively. Altogether, researchers' results indicated that all variants tested significantly impacted the activity of CYP21A2. The variants p.W202R, p.E352V, and p.R484L also impacted the protein expression (Figure 5C).
6. Kinetic Analysis of CYP21A2 Variants
The apparent kinetic constant revelated saturation for the 21OH WT with the Michaelis-Menten constant (Km) of 1.57 µM for progesterone and an apparent maximal reaction velocity (Vmax) of 0.360 nmol·min−1·mg−1 (Figure 6 and Table 4). The apparent saturation was a similar rate of the WT for p.P35L (Km 2.06 µM) and p.P433L (Km 1.91 µM), but it was six times higher for p.L199P (Km 10.24 µM), indicating that p.L199P decreases the protein affinity for the substrate progesterone. The apparent Vmax was lower than the WT for p.Pro35Leu (0.085 nmol·min−1·mg−1), p.L199P (0.252 nmol·min−1·mg−1), and p.P433L (0.055 nmol·min−1·mg−1). The apparent catalytic efficiencies of the mutated proteins were also lower than those of WT, and p.P35L had catalytic efficiency of 18% compared to the WT, p.L199P of 11%, and p.P433L of 13% (Table 5, Figure 6).
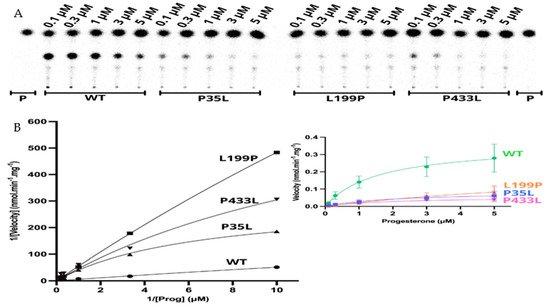
Figure 6. Kinetics assay shows a decrease in reaction velocity for three variants which have still presented activity in the functional assay. (A) Representative thin layer chromatography (TLC) from three replicates shows the CYP21A2 conversion of progesterone (Prog) (0.1, 0.3, 1, 3, and 5 µM) into 11-deoxycorticosterone (DOC). (B) The left-hand plot presents the linear plots of enzymatic activity of 21OH WT and mutations between 1/Velocity against 1/Prog (progesterone concentration) for the conversion of progesterone to DOC. Additionally, the right-hand plot presents the Michaelis Menten nonlinear curve with velocity reactions against the substrate progesterone used to obtain Vmax and Km values.
Table 5. Apparent kinetic constants and catalytic efficiency were calculated from three independent experiments.
| Wild-Type | P35L | L199P | P433L | |
|---|---|---|---|---|
| Vmax (nmol·min−1·mg−1) | 0.360 | 0.086 | 0.252 | 0.055 |
| Km (µM) | 1.57 | 2.07 | 10.24 | 1.91 |
| Vmax/Km | 0.229 | 0.041 | 0.025 | 0.029 |
Researchers' results show that the p.L199P variant, which is located close to the catalytic site, affects the substrate binding. In contrast, p.P35L and p.P433L showed inhibition of the enzyme activity through a decrease in reaction velocity.
References
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151.
- Pallan, P.S.; Wang, C.; Lei, L.; Yoshimoto, F.K.; Auchus, R.J.; Waterman, M.R.; Guengerich, F.P.; Egli, M. Human Cytochrome P450 21A2, the Major Steroid 21-Hydroxylase: Structure of the enzyme·progesterone substrate complex and rate-limiting c–h bond cleavage. J. Biol. Chem. 2015, 290, 13128–13143.
- New, M.I.; Abraham, M.; Gonzalez, B.; Dumic, M.; Razzaghy-Azar, M.; Chitayat, D.; Sun, L.; Zaidi, M.; Wilson, R.C.; Yuen, T. Genotype-phenotype correlation in 1507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc. Natl. Acad. Sci. USA 2013, 110, 2611–2616.
- Tardy, V.; Menassa, R.; Sulmont, V.; Lienhardt-Roussie, A.; Lecointre, C.; Brauner, R.; David, M.; Morel, Y. Phenotype-Genotype Correlations of 13 Rare CYP21A2 Mutations Detected in 46 Patients Affected with 21-Hydroxylase Deficiency and in One Carrier. J. Clin. Endocrinol. Metab. 2010, 95, 1288–1300.
- Janner, M.; Pandey, A.V.; Mullis, P.E.; Flück, C.E. Clinical and biochemical description of a novel CYP21A2 gene mutation 962_963insA using a new 3D model for the P450c21 protein. Eur. J. Endocrinol. 2006, 155, 143–151.
- Pandey, A.V.; Flück, C.E. NADPH P450 oxidoreductase: Structure, function, and pathology of diseases. Pharmacol. Ther. 2013, 138, 229–254.
- Ali, S.R.; Bryce, J.; Haghpanahan, H.; Lewsey, J.D.; Tan, L.E.; Atapattu, N.; Birkebaek, N.H.; Blankenstein, O.; Neumann, U.; Balsamo, A.; et al. Real-World Estimates of Adrenal Insufficiency–Related Adverse Events in Children With Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2021, 106, e192–e203.
- Carmina, E.; Dewailly, D.; Escobar-Morreale, H.F.; Kelestimur, F.; Moran, C.; Oberfield, S.; Witchel, S.F.; Azziz, R. Non-Classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: An update with a special focus on adolescent and adult women. Hum. Reprod. Update 2017, 23, 580–599.
- Krone, N.; Riepe, F.G.; Grötzinger, J.; Partsch, C.J.; Sippell, W.G. Functional characterization of two novel point mutations in the CYP21 gene causing simple virilizing forms of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin. Endocrinol. Metab. 2005, 90, 445–454.
- El-Maouche, D.; Arlt, W.; Merke, D.P. Congenital adrenal hyperplasia. Lancet 2017, 390, 2194–2210.
- Held, P.K.; Bird, I.M.; Heather, N.L. Newborn Screening for Congenital Adrenal Hyperplasia: Review of Factors Affecting Screening Accuracy. Int. J. Neonatal Screen. 2020, 6, 67.
- Flück, C.E.; Tajima, T.; Pandey, A.V.; Arlt, W.; Okuhara, K.; Verge, C.F.; Jabs, E.W.; Mendonca, B.B.; Fujieda, K.; Miller, W.L. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nat. Genet. 2004, 36, 228–230.
- Flück, C.E.; Mallet, D.; Hofer, G.; Samara-Boustani, D.; Leger, J.; Polak, M.; Morel, Y.; Pandey, A.V. Deletion of P399_E401 in NADPH cytochrome P450 oxidoreductase results in partial mixed oxidase deficiency. Biochem. Biophys. Res. Commun. 2011, 412, 572–577.
- Parween, S.; Roucher-Boulez, F.; Flück, C.E.; Lienhardt-Roussie, A.; Mallet, D.; Morel, Y.; Pandey, A.V. P450 Oxidoreductase Deficiency: Loss of Activity Caused by Protein Instability From a Novel L374H Mutation. J. Clin. Endocrinol. Metab. 2016, 101, 4789–4798.
- Fernandez-Cancio, M.; Camats, N.; Fluck, C.E.; Zalewski, A.; Dick, B.; Frey, B.M.; Monne, R.; Toran, N.; Audi, L.; Pandey, A.V. Mechanism of the Dual Activities of Human CYP17A1 and Binding to Anti-Prostate Cancer Drug Abiraterone Revealed by a Novel V366M Mutation Causing 17,20 Lyase Deficiency. Pharmaceuticals 2018, 11, 37.
- De Carvalho, D.F.; Miranda, M.C.; Gomes, L.G.; Madureira, G.; Marcondes, J.A.; Billerbeck, A.E.; Rodrigues, A.S.; Presti, P.F.; Kuperman, H.; Damiani, D.; et al. Molecular CYP21A2 diagnosis in 480 Brazilian patients with congenital adrenal hyperplasia before newborn screening introduction. Eur. J. Endocrinol. 2016, 175, 107–116.
- Lidaka, L.; Bekere, L.; Lazdane, G.; Dzivite-Krisane, I.; Kivite-Urtane, A.; Gailite, L. Non-Classical Congenital Adrenal Hyperplasia-Causing Alleles in Adolescent Girls with PCOS and in Risk Group for PCOS Development. Diagnostics 2021, 11, 980.
- Rodrigues, N.R.; Dunham, I.; Yu, C.Y.; Carroll, M.C.; Porter, R.R.; Campbell, R.D. Molecular characterization of the HLA-Linked steroid 21-hydroxylase B gene from an individual with congenital adrenal hyperplasia. EMBO J. 1987, 6, 1653–1661.
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677.
- Simonetti, L.; Bruque, C.D.; Fernández, C.S.; Benavides-Mori, B.; Delea, M.; Kolomenski, J.E.; Espeche, L.D.; Buzzalino, N.D.; Nadra, A.D.; Dain, L. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum. Mutat. 2018, 39, 5–22.
- Higashi, Y.; Tanae, A.; Inoue, H.; Hiromasa, T.; Fujii-Kuriyama, Y. Aberrant splicing and missense mutations cause steroid 21-hydroxylase deficiency in humans: Possible gene conversion products. Proc. Natl. Acad. Sci. USA 1988, 85, 7486–7490.
- Miller, W.L. Gene conversions, deletions, and polymorphisms in congenital adrenal hyperplasia. Am. J. Hum. Genet. 1988, 42, 4–7.
- Tusie-Luna, M.T.; Traktman, P.; White, P.C. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J. Biol. Chem. 1990, 265, 20916–20922.
- Barbaro, M.; Soardi, F.C.; Östberg, L.J.; Persson, B.; de Mello, M.P.; Wedell, A.; Lajic, S. In Vitro functional studies of rare CYP21A2 mutations and establishment of an activity gradient for nonclassic mutations improve phenotype prediction in congenital adrenal hyperplasia. Clin. Endocrinol. 2015, 82, 37–44.
- Haider, S.; Islam, B.; D’Atri, V.; Sgobba, M.; Poojari, C.; Sun, L.; Yuen, T.; Zaidi, M.; New, M.I. Structure-Phenotype correlations of human CYP21A2 mutations in congenital adrenal hyperplasia. Proc. Natl. Acad. Sci. USA 2013, 110, 2605–2610.
- Xu, C.; Jia, W.; Cheng, X.; Ying, H.; Chen, J.; Xu, J.; Guan, Q.; Zhou, X.; Zheng, D.; Li, G.; et al. Genotype–Phenotype correlation study and mutational and hormonal analysis in a Chinese cohort with 21-hydroxylase deficiency. Mol. Genet. Genom. Med. 2019, 7, e671.
- Wang, C.; Pallan, P.S.; Zhang, W.; Lei, L.; Yoshimoto, F.K.; Waterman, M.R.; Egli, M.; Guengerich, F.P. Functional analysis of human cytochrome P450 21A2 variants involved in congenital adrenal hyperplasia. J. Biol. Chem. 2017, 292, 10767–10778.
- Coeli, F.B.; Soardi, F.C.; Bernardi, R.D.; De Araújo, M.; Paulino, L.C.; Lau, I.F.; Petroli, R.J.; De Lemos-Marini, S.H.; Baptista, M.T.; Guerra-Júnior, G.; et al. Novel deletion alleles carrying CYP21A1P/A2chimeric genes in Brazilian patients with 21-hydroxylase deficiency. BMC Med. Genet. 2010, 11, 104.
- Silveira, E.L.; Elnecave, R.H.; dos Santos, E.P.; Moura, V.; Pinto, E.M.; van der Linden Nader, I.; Mendonca, B.B.; Bachega, T.A. Molecular analysis of CYP21A2 can optimize the follow-up of positive results in newborn screening for congenital adrenal hyperplasia. Clin. Genet. 2009, 76, 503–510.
- Santos-Silva, R.; Cardoso, R.; Lopes, L.; Fonseca, M.; Espada, F.; Sampaio, L.; Brandão, C.; Antunes, A.; Bragança, G.; Coelho, R. CYP21A2 Gene Pathogenic Variants: A Multicenter Study on Genotype–Phenotype Correlation from a Portuguese Pediatric Cohort. Horm. Res. Paediatr. 2019, 91, 33–45.
- Carvalho, B.; Pereira, M.; Marques, C.J.; Carvalho, D.; Leão, M.; Oliveira, J.P.; Barros, A.; Carvalho, F. Comprehensive genetic analysis and structural characterization of CYP21A2 mutations in CAH patients. Exp. Clin. Endocrinol. Diabetes 2012, 120, 535–539.
More
Information
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
29 Mar 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No