Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shujing Xu | + 1652 word(s) | 1652 | 2022-02-09 05:18:31 | | | |
| 2 | Jason Zhu | -45 word(s) | 1607 | 2022-02-16 03:11:04 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Xu, S. Newly Emerging Antiviral Strategies. Encyclopedia. Available online: https://encyclopedia.pub/entry/19478 (accessed on 09 August 2026).
Xu S. Newly Emerging Antiviral Strategies. Encyclopedia. Available at: https://encyclopedia.pub/entry/19478. Accessed August 09, 2026.
Xu, Shujing. "Newly Emerging Antiviral Strategies" Encyclopedia, https://encyclopedia.pub/entry/19478 (accessed August 09, 2026).
Xu, S. (2022, February 16). Newly Emerging Antiviral Strategies. In Encyclopedia. https://encyclopedia.pub/entry/19478
Xu, Shujing. "Newly Emerging Antiviral Strategies." Encyclopedia. Web. 16 February, 2022.
Copy Citation
Various modern innovative methods or integrated paradigms are now being applied to drug discovery for significant resistance in order to simplify the drug process. In recent years, several strategies have been employed to discover novel antiviral agents with new scaffolds and better resistance profiles, including proteolysis targeting chimera (PROTAC), ribonuclease targeting chimera (RIBOTAC), targeted covalent inhibitors, and topology-matching design.
viruses
antiviral drugs
medicinal chemistry strategies
1. Proteolysis Targeting Chimera (PROTAC)
PROTACs have become an emerging drug discovery paradigm to target proteins through promoting and realizing the degradation of target proteins via the ubiquitin–proteasome system (UPS) [1]. PROTACs are hetero-bifunctional molecules consisting of a ligand for the protein of interest (POI), an E3 ubiquitin ligase recruitment ligand and a linker. Bifunctional PROTAC molecules bind to the POI with one end, while the other end binds to an E3 ligase to shorten the distance between them in vivo. The E3 ligase then mediates the transfer of ubiquitin from an E2 enzyme to the POI, and finally the ubiquitylated POI is knocked down by the proteasome [2][3].
In 2019, Yang et al. [4] reported a PROTAC molecule that could degrade the hepatitis C virus (HCV) protease. Telaprevir (1), a reversible covalent inhibitor binding to the active site of HCV protease, the cocrystal structure of telaprevir in complex with the viral protease, showed that the solvent-exposed pyrazine ring could be derivatized with different linkers conjugated to chemical binders of cereblon (CRBN), the substrate receptor for the CUL4–RBX1–DDB1–CRBN E3 ubiquitin ligase complex (CRL4CRBN). Therefore, telaprevir could serve as a protein–ligand, being conjugated to ligands that recruit the CRL4CRBN ligase complex, producing compounds that could both inhibit and induce the degradation of the HCV NS3/4A protease. The CRBN-binding moieties of the PROTAC molecules were derived from lenalidomide, pomalidomide or a tricyclic imide moiety. An optimized one, DGY-08-097 (2, Figure 1), effectively inhibited HCV in a cellular infection model (EC50 = 748 nM), which proved that the degradation of protein was helpful to its antiviral activity. Finally, the researchers concluded that these new types of antiviral agents could overcome viral variation and thereby solve drug-resistance to traditional enzymatic inhibitors. The finding confirmed that these small-molecule degraders were less vulnerable to mutations that affected ligand binding and could be employed to inhibit or treat viral variants associated with resistance to traditional inhibitors.
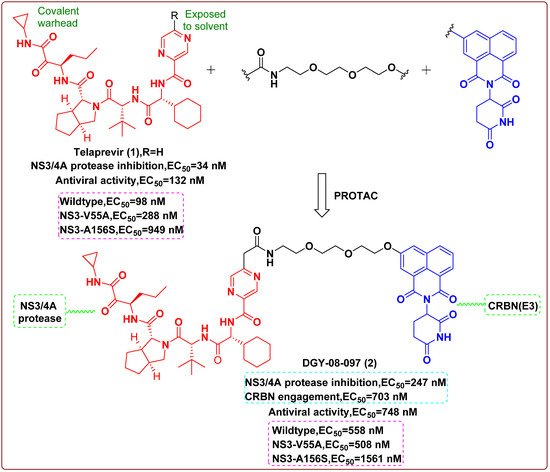
Figure 1. Chemical structures and inhibitory activities of telaprevir (1) and the degrader derivative DGY-08-097 (2) [4].
2. Ribonuclease Targeting Chimera (RIBOTAC)
RIBOTAC is a new strategy for RNA degradation. RIBOTAC includes an RNA-binding small molecule and a ribonuclease (RNase) L-recruiting module aiming to degrade the viral genome [5][6]. RNase L acts in innate immunity and is expressed at minute levels as an inactive monomer in all cells, which is activated and dimerized during viral infection with inherent substrate specificity [7]. RIBOTACs locally recruit RNase L to the expected target to achieve the effect of selective cleavage.
In 2020, Haniff et al. [8] designed multiple bioactive small molecules targeting a functional structure within the RNA genome of SARS-CoV-2. An analysis of RNA genome structure afforded a modified model of the SARS-CoV-2 frameshifting element (FSE), especially its attenuator hairpin, which controlled the translation of pp1a and pp1ab polyproteins that were essential for viral replication and pathogenesis. Using Absorb Array and luciferase reporter-based cellular assays, they identified a drug-like small molecule (C5, 3) that selectively bound to and stabilized the revised attenuator hairpin structure of FSE with a Kd of 11 nM, reducing its frameshifting efficiency in cells. The ligand was further elaborated into an RNA degrader (C5-RIBOTAC, 4) to recruit a cellular ribonuclease to destroy the viral genome that validated direct target engagement and enhanced antiviral potency via targeted degradation of the viral RNA. Eventually, the RIBOTAC-based lead optimization strategy enhanced the antiviral activity of the lead compound at least 10-fold.
3. Targeted Covalent Inhibitors
The development of structural biology and bioinformatics has greatly promoted the rational design of targeted covalent inhibitors (TCIs). Covalent inhibitors can interact with specific target proteins to form covalent bonds that result in changes in the conformation of proteins, thus interfering with the normal function of the protein [9]. The covalent binding with the target can be divided into two related but discontinuous processes: (i) the inhibitor reversibly binds to the target, making the functional groups on the weak electrophilic ligands adjacent to the specific nucleophilic residues on the protein; (ii) the ligand reacts with the functional groups involved in the protein to form a covalent bond [10][11]. In recent years, TCIs have received growing attention from the antiviral field due to their significant advances in terms of efficacy and selectivity.
Resistance related to the Tyr181Cys (Y181C) mutation in HIV-1 reverse transcriptase (RT) is one of the main obstacles for the development of nonnucleoside RT inhibitors (NNRTIs). In 2017, Chan et al. [12] reported covalent inhibitors of Y181C RT that could completely knock out activity of the resistant mutant. Enzyme inhibition kinetics, mass spectrometry, protein crystallography and antiviral activity detection provided compelling evidence for covalent modification of Cys181. Success was obtained for the chloromethylamide 5 and the acrylamide 6, and they could form covalent bonds with the sulfhydryl group of Cys181; it may be possible to dose them less frequently than noncovalent inhibitors (Figure 2). It was the first time that an irreversible covalent inhibition strategy was successfully applied to HIV-1 RT; diversity-oriented warhead selection made it possible to systematically explore chemical space.

Figure 2. (A) Chemical structure of 5 and crystal structures of Y181C RT in complex with 5 (PDB code: 5VQX); 5 forms a covalent bond with the sulfhydryl group of Cys181. (B) The same as A but with Y181C:6 (PDB code: 5VQV) [12]. The figures were generated by PyMol.
In 2020, Hoffman et al. [13] reported the discovery and characterization of a potent ketone-based covalent inhibitor of SARS-CoV-2 coronavirus 3CL protease (3CLpro). 3CLpro, as the main protease, is critical for mediating viral replication and transcription. The hydroxymethylketone derivative 7 exhibited potent SARS-CoV inhibition in 3CLpro and antiviral assays. Cocrystal structures of 7 in complex with 3CLpro of SARS-CoV-2 confirmed that the warhead hydroxymethylketone carbonyl carbon of 7 formed a covalent bond to the sulfur of the Cys145 in 3CLpro active-site, producing a tetrahedral carbinol complex. This carbinol hydroxyl formed hydrogen bonds with the backbone NH of Cys145 and with the amide NH of Gly143 via a bridging water molecule. Another key active-site interaction was the hydrogen bond between the primary alcohol moiety of 7 and the catalytic His41 (Figure 3A). Additionally, 7 displayed acceptable solubility, stability in plasma and low in vitro and in vivo clearances, which were suitable for further development as an anti-SARS-CoV-2 drug candidate. Moreover, Dai et al. [14] also reported two potent inhibitors (8 and 9, Figure 3B) that were covalently bound to Cys145 of 3CLpro. Both of them showed good pharmacokinetic properties in vivo, and 8 also exhibited low toxicity, suggesting that these compounds are promising anti-SARS-CoV-2 drug candidates.

Figure 3. (A) Chemical structure of 7 and the schematic rendering of the active site with dashed lines represented as hydrogen bonds with key residues and curved lines to show S1 and S2 binding pockets [13]; (B) chemical structures of 8 and 9; inhibitory activities against SARS-CoV-2 3CLPro(IC50) and in vitro inhibition of 3CLPro(EC50) [14].
4. Topology-Matching Design
Influenza A virus (IAV) is an enveloped RNA virus, of which the membrane anchors two viral proteins that regulate interactions between the virion and host cells, namely hemagglutinin (HA) and neuraminidase (NA) [15]. From a topological viewpoint, the virion of IAV is a nanosized particle of about 100 nm with a spiky surface created by the HA and NA [16]. For nano-inhibitors, it is crucial to match the size and topology of the virion in order to achieve competitive binding with the virus/cell interaction.
In 2020, Nie et al. [17] demonstrated the concept of “topology-matching design” for virus inhibitors. They designed a nano-inhibitor with a nano-topological structure matching the IAV virions and showed hetero-multivalent inhibitory effects on HA and NA (Figure 4A). The synthesized nano-inhibitor could neutralize the viral particle extracellularly and block its attachment and enter host cells. The virus replication was substantially reduced by six orders of magnitude, reaching more than 99.999% inhibition even after 24 h of infection, which demonstrated that such a nano-inhibitor might be a potent anti-influenza agent. Moreover, they also found a spiky nano-inhibitor with matched topography to IAV virions. Due to the short spikes inserted into the glycoprotein gap of the IAV virion, the binding of the nanostructures with spikes between 5 and 10 nm was substantially better than that of smooth nanoparticles (Figure 4B). In addition, targeting IAV by an erythrocyte membrane (EM) could efficiently prevent IAV virion binding to the cells and inhibit subsequent infection.

Figure 4. (A) Proposed binding patterns between nano-inhibitor and IAV particles [17]; (B) proposed binding patterns between spiky nanoparticle-based inhibitor and IAV particles [18]; (C) proposed binding patterns between IAV and the heteromultivalent nanobowl (Hetero-MNB), where sialic acid and zanamivir bind to HA and NA, respectively, and the bowl shape facilitating the capping to the surface of the virus particle [19].
In 2021, the same group reported heteromultivalent topology-matched nanostructures as effective and broad-spectrum IAV inhibitors. The heteromultivalent binding moieties were transferred to bowl-like nanostructures that matched the spherical surface of the virus, coating the inhibitor surface with a cell-derived membrane as a native source of sialic acids and complementing the cell-derived membrane with zanamivir to increase the IAV–membrane interaction by heteromultivalent binding (Figure 4C). Unlike the traditional homomultivalent inhibitors, the IC50 value of the heteromultivalent inhibitors was 32.4 ± 13.7 ug/mL owing to the synergistic multivalent effects and the topology-matched shape. The virus propagation was reduced by more than 99.99% at a dose that did not cause cytotoxicity. Since multiple binding sites have also been identified on the S protein of SARS-CoV-2, it is envisaged that heteromultivalent nanostructures may also be employed in seeking effective SARS-CoV-2 inhibitors [19].
References
- Lai, A.C.; Crews, C.M. Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 2017, 16, 101–114.
- Paiva, S.L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119.
- Li, X.; Song, Y. Proteolysis-targeting chimera (PROTAC) for targeted protein degradation and cancer therapy. J. Hematol. Oncol. 2020, 13, 50.
- de Wispelaere, M.; Du, G.; Donovan, K.A.; Zhang, T.; Eleuteri, N.A.; Yuan, J.C.; Kalabathula, J.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; et al. Small molecule degraders of the hepatitis C virus protease reduce susceptibility to resistance mutations. Nat. Commun. 2019, 10, 3468.
- Costales, M.G.; Aikawa, H.; Li, Y.; Childs-Disney, J.L.; Abegg, D.; Hoch, D.G.; Pradeep Velagapudi, S.; Nakai, Y.; Khan, T.; Wang, K.W.; et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 2406–2411.
- Costales, M.G.; Suresh, B.; Vishnu, K.; Disney, M.D. Targeted Degradation of a Hypoxia-Associated Non-coding RNA Enhances the Selectivity of a Small Molecule Interacting with RNA. Cell Chem. Biol. 2019, 26, 1180–1186.e1185.
- Costales, M.G.; Matsumoto, Y.; Velagapudi, S.P.; Disney, M.D. Small Molecule Targeted Recruitment of a Nuclease to RNA. J. Am. Chem. Soc. 2018, 140, 6741–6744.
- Haniff, H.S.; Tong, Y.; Liu, X.; Chen, J.L.; Suresh, B.M.; Andrews, R.J.; Peterson, J.M.; O’Leary, C.A.; Benhamou, R.I.; Moss, W.N.; et al. Targeting the SARS-CoV-2 RNA Genome with Small Molecule Binders and Ribonuclease Targeting Chimera (RIBOTAC) Degraders. ACS Cent. Sci. 2020, 6, 1713–1721.
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 2015, 20, 1061–1073.
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724.
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830.
- Chan, A.H.; Lee, W.G.; Spasov, K.A.; Cisneros, J.A.; Kudalkar, S.N.; Petrova, Z.O.; Buckingham, A.B.; Anderson, K.S.; Jorgensen, W.L. Covalent inhibitors for eradication of drug-resistant HIV-1 reverse transcriptase: From design to protein crystallography. Proc. Natl. Acad. Sci. USA 2017, 114, 9725–9730.
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747.
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335.
- Calder, L.J.; Rosenthal, P.B. Cryomicroscopy provides structural snapshots of influenza virus membrane fusion. Nat. Struct. Mol. Biol. 2016, 23, 853–858.
- Amaro, R.E.; Ieong, P.U.; Huber, G.; Dommer, A.; Steven, A.C.; Bush, R.M.; Durrant, J.D.; Votapka, L.W. A Computational Assay that Explores the Hemagglutinin/Neuraminidase Functional Balance Reveals the Neuraminidase Secondary Site as a Novel Anti-Influenza Target. ACS Cent. Sci. 2018, 4, 1570–1577.
- Nie, C.; Parshad, B.; Bhatia, S.; Cheng, C.; Stadtmuller, M.; Oehrl, A.; Kerkhoff, Y.; Wolff, T.; Haag, R. Topology-Matching Design of an Influenza-Neutralizing Spiky Nanoparticle-Based Inhibitor with a Dual Mode of Action. Angew. Chem. Int. Ed. Engl. 2020, 59, 15532–15536.
- Nie, C.; Stadtmuller, M.; Yang, H.; Xia, Y.; Wolff, T.; Cheng, C.; Haag, R. Spiky Nanostructures with Geometry-matching Topography for Virus Inhibition. Nano. Lett. 2020, 20, 5367–5375.
- Nie, C.; Stadtmüller, M.; Parshad, B.; Wallert, M.; Ahmadi, V.; Kerkhoff, Y.; Bhatia, S.; Block, S.; Cheng, C.; Wolff, T.; et al. Heteromultivalent topology-matched nanostructures as potent and broad-spectrum influenza A virus inhibitors. Sci. Adv. 2021, 7, eabd3803.
More
Information
Subjects:
Allergy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
940
Revisions:
2 times
(View History)
Update Date:
16 Feb 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No