 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kathlyn Laval | + 3844 word(s) | 3844 | 2022-01-28 07:45:40 | | | |
| 2 | Peter Tang | -67 word(s) | 3777 | 2022-02-14 07:19:13 | | | | |
| 3 | Lindsay Dong | Meta information modification | 3777 | 2022-03-28 04:43:15 | | |
Video Upload Options
Pseudorabies virus (PRV) is an alphaherpesvirus related to varicella-zoster virus (VZV) and herpes simplex virus type 1 (HSV1). PRV is the causative agent of Aujeskzy’s disease in swine. PRV infects mucosal epithelium and the peripheral nervous system (PNS) of its host where it can establish a quiescent, latent infection. While the natural host of PRV is the swine, a broad spectrum of mammals, including rodents, cats, dogs, and cattle can be infected. Since the nineteenth century, PRV infection is known to cause a severe acute neuropathy, the so called “mad itch” in non-natural hosts, but surprisingly not in swine. In the past, most scientific efforts have been directed to eradicating PRV from pig farms by the use of effective marker vaccines, but little attention has been given to the processes leading to the mad itch.
1. History of Aujeszky’s Disease
2. Pseudorabies Virus (PRV)
3. The pathogenesis of PRV-induced neuropathic itch
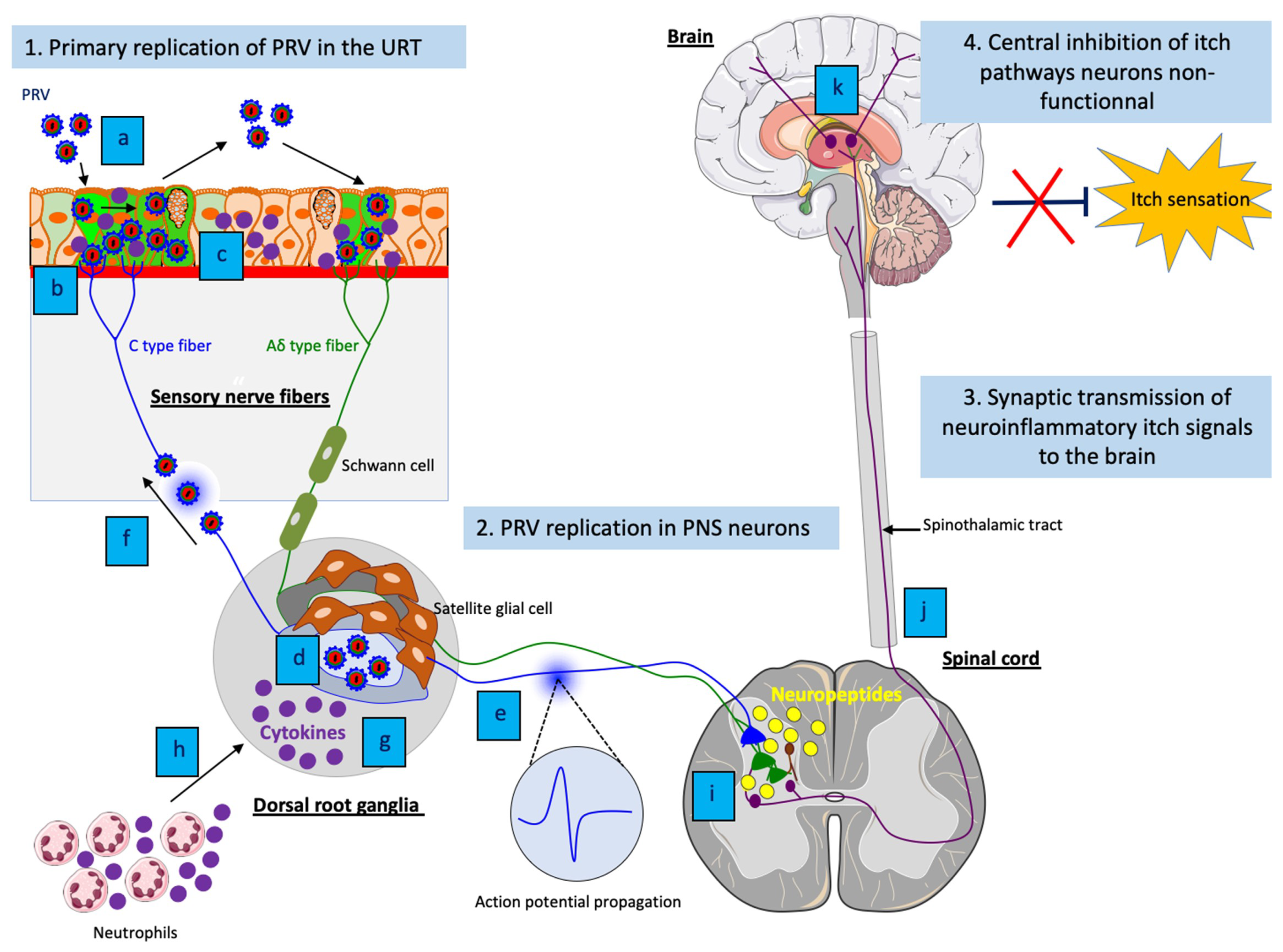
PRV is a highly contagious pathogen that causes respiratory disease, neurological disorders and abortion in swine. Transmission occurs primarily through direct contact with oral and nasal secretions. Other animal species (non-natural hosts) are commonly infected through direct contact with pigs or through the consumption of raw offal from infected pigs. While PRV-infected swine do not itch and can survive the infection, non-natural hosts infected with the virus develop a severe pruritus and usually die within 2 days. In these animals, PRV enters nerve endings of the PNS that innervate the nasal mucosa of the upper respiratory tract and initiates a productive infection in PNS neurons. After replication in the PNS, progeny virions may spread in the retrograde direction from the PNS to the CNS if the animals survive long enough (Figure 1) [26].
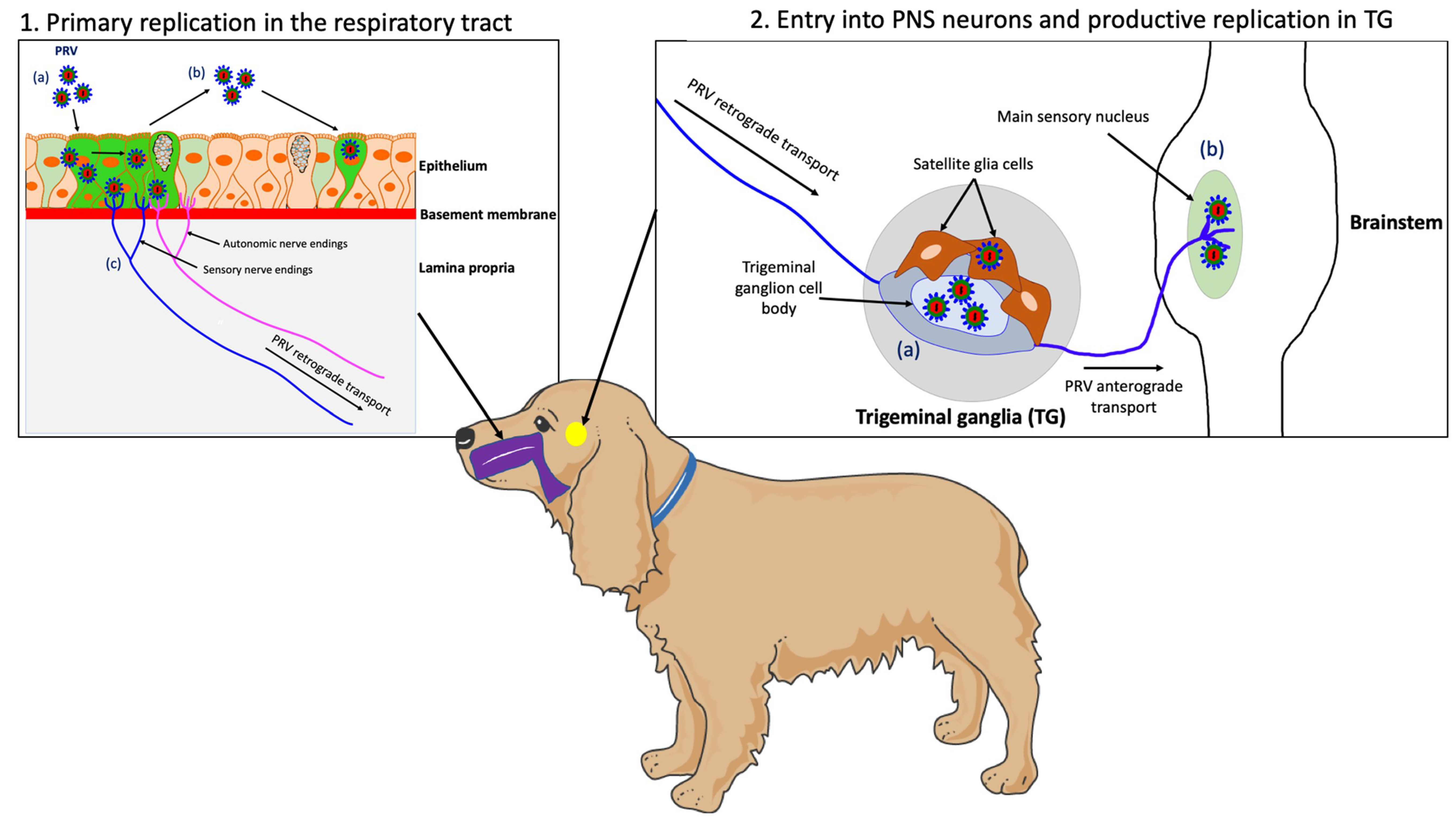
Figure 1. Schematic representation of the pathogenesis of PRV in a non-natural host, the dog. (1) PRV first replicates in the epithelium of the upper respiratory tract; (a) PRV infection; (b) Viral spread within the respiratory epithelium and viral shedding; (c) PRV enters nerve endings of the PNS, including those coming from the trigeminal ganglia (TG) and spreads in the retrograde direction to the ganglia. (2) PRV initiates a productive infection in TG neurons; (a) PRV replicates in cell bodies of TG neurons; (b) New progeny virions can further spread in the anterograde direction and infect the CNS (brainstem). Purple = respiratory tract; yellow = TG.
4. PRV infection in mice: a new animal model for VZV-induced peripheral neuropathies
A better understanding of PRV-induced neuroinflammatory responses in mice may provide new insights in the initiation and development of virus-induced neuroinflammation during other herpesvirus infections. For instance, this animal model could be useful to dissect the mechanisms of neuropathic itch in patients with post-herpetic lesions (e.g. herpes zoster (HZ); shingles). Indeed, the neuropathogenesis and immunopathogenesis of VZV and PRV infections are remarkably similar. Reactivation of VZV causes a self-limited dermatomal rash with pain and itching, which is accompanied by inflammation of the skin. The HZ lesions can be reduced by treatment with antivirals [49]. However, postherpetic neuralgia (PHN) and postherpetic itch (PHI) are two common complications of HZ that can occur in some cases in up to 50% of patients with shingles [50].
PHN consists of a burning and stabbing pain while PHI is characterized by a relentless itch in the same area of the HZ rash, resulting in serious injuries due to scratching. Both PHN and PHI can last for months or years after resolution of the HZ rash, thus severely impacting the life quality of infected people. Antiviral therapy for acute HZ does not eliminate the risk of PHN, and no beneficial effect of any antiviral drug on established PHN has been shown. It was suggested that PHN is caused by VZV-induced inflammation and axonal damage, which gives rise to hyperexcitability, marked by spontaneous firing of PNS neurons. These neurons may have a lower excitation threshold to pain, thus causing neuropathic pain [51]. In contrast, the underlying mechanism(s) of PHI is largely unknown. Despite some similarities between itch and pain pathways, treatments against pain are not efficient in relieving itch. Current treatments against neuropathic itch are very limited and lack specificity, and for many patients with PHI no alleviation can be provided [52].
The narrow host range and lack of clinical disease have limited the use of animal models to investigate the pathogenesis of VZV infection [53]. So far, investigation of PHI has been limited by the lack of a relevant in vivo neuropathic itch animal model. Interestingly, VZV and PRV infections present multiple similarities in genome sequence, clinical signs, pathogenesis and immunity. At the level of innate immunity, the exact same concentration of IL-6 (∼30,000 pg/ml) has been demonstrated in both VZV-infected human explants and PRV-infected mouse footpad by ELISA [54][45]. Since VZV does not productively replicate in rodents, PRV-induced neuropathic itch in mice may represent a promising model to further understand the pathogenesis of PHI caused by VZV infection.
5. Conclusions
Since the first case of mad itch was described 207 years ago, the characteristic pruritus caused by PRV infection in non-natural hosts has been frequently reported throughout the years. Currently, relatively few studies have focused on this particular aspect of PRV pathogenesis. This paucity of information is mainly because PRV remained a major viral disease in swine, causing substantial economic losses to pig producers. Therefore, the efforts of the research community were primarily focused on developing effective vaccines aimed at eradication of the virus. Recently, researchers dissected the molecular and cellular mechanisms of PRV-induced neuropathic itch using several mouse models and emphasized the innate immune response as a central player. Good control of the inflammatory response during PRV infection of swine likely prevents the neuropathic pruritus experienced by infected non-natural hosts. Most importantly, PRV infection of mice has proven to be a suitable animal model to study PRV-induced neuropathic itch. This animal model may also provide useful insights into the pathogenesis of other herpesvirus infections, such as those following VZV infection. The model may lead to the development of innovative therapeutic strategies.[9]
References
- A. Aujeszky; Uber eine neue Infektion krankheit bei Haustieren. Zbl Bakt Abt Orig 1902, 32, 353-357.
- J. Schmiedhoffer; Beiträge zur Pathologieder infektiösen Bulbär paralyse (Aujeszky-schen Krankheit). Z Infekt Krankh Parasit Hyg Haustier 1910, 8, 383-405.
- R.E. Shope; An experimental study of ‘mad itch’ with especial reference to its relationship to pseudorabies. Journal of Experimental Medicine 1931, 54, 233-248.
- R.E. Shope; Pseudorabies is a contagious disease in swine. Science 1934, 80, 102-103.
- A.B. Sabin; The immunological identity of a virus isolated from a human case of ascending myelitis associated with visceral necrosis. Journal of Experimental Pathology 1934, 15, 248-268.
- A.B. Sabin; Acute ascending myelitis following monkey bite with isolation of a virus capable of reproducing disease. Journal of Experimental Medicine 1934, 59, 115.
- A.B. Sabin; Progression of different nasally installed viruses along different nervous pathways in the same host. Proceedings of the Society for Experimental Biology and Medicine 1938, 38, 270-275.
- I. Steiner; P.G.E. Kennedy; A.R. Pachner; The neurotropic herpes viruses: herpes simplex and varicella-zoster. The Lancet Neurology 2007, 6, 1015-1028.
- Barbara G. Klupp; Christoph J. Hengartner; Thomas C. Mettenleiter; Lynn W. Enquist; Complete, Annotated Sequence of the Pseudorabies Virus Genome. Journal of Virology 2004, 78, 424-440, 10.1128/jvi.78.1.424-440.2004.
- T.C. Mettenleiter; Herpesvirus assembly and egress. Journal of Virology 2002, 76, 1537-1547.
- Lisa E. Pomeranz; Ashley E. Reynolds; Christoph J. Hengartner; Molecular Biology of Pseudorabies Virus: Impact on Neurovirology and Veterinary Medicine. Microbiology and Molecular Biology Reviews 2005, 69, 462-500, 10.1128/mmbr.69.3.462-500.2005.
- Akihiko Ikoma; Martin Steinhoff; Sonja Ständer; Gil Yosipovitch; Martin Schmelz; The neurobiology of itch. Nature Reviews Neuroscience 2006, 7, 535-547, 10.1038/nrn1950.
- Dustin Green; Xinzhong Dong; The cell biology of acute itch. Journal of Cell Biology 2016, 213, 155-161, 10.1083/jcb.201603042.
- Gil Yosipovitch; Alan B Fleischer; Itch Associated with Skin Disease. American Journal of Clinical Dermatology 2003, 4, 617-622, 10.2165/00128071-200304090-00004.
- A. Galatian; G. Stearns; R. Grau; Pruritus in connective tissue and other common systemic disease states. Cutis 2009, 84, 207-214.
- Andreas Binder; Jana Koroschetz; Ralf Baron; Disease mechanisms in neuropathic itch. Nature Clinical Practice Cardiovascular Medicine 2008, 4, 329-337, 10.1038/ncpneuro0806.
- Gil Yosipovitch; Lena S Samuel; Neuropathic and psychogenic itch. Dermatologic Therapy 2008, 21, 32-41, 10.1111/j.1529-8019.2008.00167.x.
- Anne Louise Oaklander; Neuropathic Itch. Seminars in Cutaneous Medicine and Surgery 2011, 30, 87-92, 10.1016/j.sder.2011.04.006.
- M. Ringkamp; R. Meyer. Frontiers in Neuroscience Pruriceptors; E. Carstens and T. Akiyama, Eds.; in Itch: Mechanisms and Treatment: CRC Press/Taylor & Francis(c), LLC.: Boca Raton (FL), 2014; pp. N/A.
- B.A. Robbins; G.J. Scmieder. Brachioradial pruritus; StatPearls, Eds.; StatPearls: StatPearls Publishing StatPearls Publishing LLC.: Treasure Island (FL)., 2020; pp. N/A.
- Asit Mittal; Ankita Srivastava; Manisha Balai; Ashok Kumar Khare; A study of postherpetic pruritus. Indian Dermatology Online Journal 2016, 7, 343-344, 10.4103/2229-5178.185479.
- M.C. Koeppel; C. Bramont; M. Ceccaldi; M. Habib; J. Sayag; Paroxysmal pruritus and multiple sclerosis. British Journal of Dermatology 1993, 129, 597-598, 10.1111/j.1365-2133.1993.tb00492.x.
- Martin Steinhoff; Martin Schmelz; Imre Lőrinc Szabó; Anne Louise Oaklander; Clinical presentation, management, and pathophysiology of neuropathic itch. The Lancet Neurology 2018, 17, 709-720, 10.1016/s1474-4422(18)30217-5.
- Steve Davidson; Xijing Zhang; Sergey G Khasabov; Donald A Simone; Glenn J Giesler Jr; Relief of itch by scratching: state-dependent inhibition of primate spinothalamic tract neurons. Nature Neuroscience 2009, 12, 544-546, 10.1038/nn.2292.
- Gil Yosipovitch; Yozo Ishiuji; Tejesh S. Patel; Maria Isabel Hicks; Yoshitetsu Oshiro; Robert A. Kraft; Erica Winnicki; Robert C. Coghill; The Brain Processing of Scratching. Journal of Investigative Dermatology 2008, 128, 1806-1811, 10.1038/jid.2008.3.
- Kathlyn Laval; Lynn W. Enquist; The Neuropathic Itch Caused by Pseudorabies Virus. Pathogens 2020, 9, 254, 10.3390/pathogens9040254.
- John Dempsher; Martin G. Larrabee; Frederik B. Bang; David Bodian; Physiological Changes in Sympathetic Ganglia Infected With Pseudorabies Virus. American Journal of Physiology-Legacy Content 1955, 182, 203-216, 10.1152/ajplegacy.1955.182.1.203.
- G.S. Liao; M. Maillard; M. Kiraly; Ion channels involved in the presynaptic hyperexcitability induced by herpes virus suis in rat superior cervical ganglion. Neuroscience 1991, 41, 797-807, 10.1016/0306-4522(91)90370-4.
- T. Tokumaru; Pseudorabies virus - induced neural hyperreactivity following occular and skin infections in the rat. Research communications in chemical pathology and pharmacology 1975, 10, 533-542.
- Kelly M. McCarthy; David W. Tank; Lynn W. Enquist; Pseudorabies Virus Infection Alters Neuronal Activity and Connectivity In Vitro. PLOS Pathogens 2009, 5, e1000640, 10.1371/journal.ppat.1000640.
- Herman Favoreel; Geert Van Minnebruggen; Hans J. Nauwynck; Lynn W. Enquist; Maurice B. Pensaert; A Tyrosine-Based Motif in the Cytoplasmic Tail of Pseudorabies Virus Glycoprotein B Is Important for both Antibody-Induced Internalization of Viral Glycoproteins and Efficient Cell-to-Cell Spread. Journal of Virology 2002, 76, 6845-6851, 10.1128/jvi.76.13.6845-6851.2002.
- Andrea E. Granstedt; Jens Bernhard Bosse; Stephan Thiberge; Lynn W. Enquist; In vivo imaging of alphaherpesvirus infection reveals synchronized activity dependent on axonal sorting of viral proteins. Proceedings of the National Academy of Sciences 2013, 110, E3516-E3525, 10.1073/pnas.1311062110.
- A. D. Brideau; J. P. Card; L. W. Enquist; Role of Pseudorabies Virus Us9, a Type II Membrane Protein, in Infection of Tissue Culture Cells and the Rat Nervous System. Journal of Virology 2000, 74, 834-845, 10.1128/jvi.74.2.834-845.2000.
- K. Laval; J. B. Vernejoul; J. Van Cleemput; O. O. Koyuncu; L. W. Enquist; Virulent Pseudorabies Virus Infection Induces a Specific and Lethal Systemic Inflammatory Response in Mice. Journal of Virology 2018, 92, e01614-18, 10.1128/jvi.01614-18.
- Elizabeth E. Brittle; Ashley E. Reynolds; L. W. Enquist; Two Modes of Pseudorabies Virus Neuroinvasion and Lethality in Mice. Journal of Virology 2004, 78, 12951-12963, 10.1128/jvi.78.23.12951-12963.2004.
- Paul J. Husak; Timothy Kuo; L. W. Enquist; Pseudorabies Virus Membrane Proteins gI and gE Facilitate Anterograde Spread of Infection in Projection- Specific Neurons in the Rat. Journal of Virology 2000, 74, 10975-10983, 10.1128/jvi.74.23.10975-10983.2000.
- M. Yang; J. P. Card; R. S. Tirabassi; R. R. Miselis; L. W. Enquist; Retrograde, Transneuronal Spread of Pseudorabies Virus in Defined Neuronal Circuitry of the Rat Brain Is Facilitated by gE Mutations That Reduce Virulence. Journal of Virology 1999, 73, 4350-4359, 10.1128/jvi.73.5.4350-4359.1999.
- Martin Schmelz; Itch Processing in the Skin.. Frontiers in Medicine 2019, 6, 167, 10.3389/fmed.2019.00167.
- Smriti Iyengar; Michael H. Ossipov; Kirk W. Johnson; The role of calcitonin gene–related peptide in peripheral and central pain mechanisms including migraine. Pain 2017, 158, 543-559, 10.1097/j.pain.0000000000000831.
- Xue Feng Wang; Tong Tong Ge; Jie Fan; Wei Yang; Bingjin Li; Ran Ji Cui; The role of substance P in epilepsy and seizure disorders. Oncotarget 2017, 8, 78225-78233, 10.18632/oncotarget.20606.
- Niklaus H. Mueller; Donald H. Gilden; Randall J. Cohrs; Ravi Mahalingam; Maria A. Nagel; Varicella Zoster Virus Infection: Clinical Features, Molecular Pathogenesis of Disease, and Latency. Neurologic Clinics 2008, 26, 675-697, 10.1016/j.ncl.2008.03.011.
- Toshio Tanaka; Masashi Narazaki; Tadamitsu Kishimoto; IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harbor Perspectives in Biology 2014, 6, a016295-a016295, 10.1101/cshperspect.a016295.
- Kwee Yong; Granulocyte colony‐stimulating factor (G‐CSF) increases neutrophil migration across vascular endothelium independent of an effect on adhesion: comparison with granulocyte‐macrophage colony‐stimulating factor (GM‐CSF). British Journal of Haematology 1996, 94, 40-47, 10.1046/j.1365-2141.1996.d01-1752.x.
- S.K. Shaw; S.A. Owolabi; J. Bagley; N. Morin; E. Cheng; B.W. LeBlanc; M. Kim; P. Harty; S.G. Waxman; C.Y. Saab; et al. Activated polymorphonuclear cells promote injury and excitability of dorsal root ganglia neurons. Experimental Neurology 2008, 210, 286-294, 10.1016/j.expneurol.2007.11.024.
- Kathlyn Laval; Jolien Van Cleemput; Jonah B. Vernejoul; Lynn W. Enquist; Alphaherpesvirus infection of mice primes PNS neurons to an inflammatory state regulated by TLR2 and type I IFN signaling. PLOS Pathogens 2019, 15, e1008087, 10.1371/journal.ppat.1008087.
- Kathlyn Laval; Carola J. Maturana; Lynn W. Enquist; Mouse Footpad Inoculation Model to Study Viral-Induced Neuroinflammatory Responses. Journal of Visualized Experiments 2020, N/A, e61121, 10.3791/61121.
- Carmen D. Rietdijk; Richard J. A. Van Wezel; Johan Garssen; Aletta D. Kraneveld; Neuronal toll-like receptors and neuro-immunity in Parkinson's disease, Alzheimer's disease and stroke. Neuroimmunology and Neuroinflammation 2016, 3, 27, 10.20517/2347-8659.2015.28.
- Donghoon Kim; Myung Ah Kim; Ik-Hyun Cho; Mi Sun Kim; Soojin Lee; Eun-Kyeong Jo; Se-Young Choi; Kyungpyo Park; Joong Soo Kim; Shizuo Akira; et al.Heung Sik NaSeog Bae OhSung Joong Lee A Critical Role of Toll-like Receptor 2 in Nerve Injury-induced Spinal Cord Glial Cell Activation and Pain Hypersensitivity. Journal of Biological Chemistry 2007, 282, 14975-14983, 10.1074/jbc.m607277200.
- Jr. John W. Gnann; Varicella‐Zoster Virus: Atypical Presentations and Unusual Complications. The Journal of Infectious Diseases 2002, 186, S91-S98, 10.1086/342963.
- Rhonda G. Kost; Stephen E. Straus; Postherpetic Neuralgia — Pathogenesis, Treatment, and Prevention. New England Journal of Medicine 1996, 335, 32-42, 10.1056/nejm199607043350107.
- Gary J Bennett; C Peter N Watson; Herpes Zoster and Postherpetic Neuralgia: Past, Present and Future. Pain Research and Management 2009, 14, 275-282, 10.1155/2009/380384.
- Valentina Semionov; Pesach Shvartzman; Post Herpetic Itching—A Treatment Dilemma. The Clinical Journal of Pain 2008, 24, 366-368, 10.1097/ajp.0b013e3181633fb1.
- Kristen Haberthur; Ilhem Messaoudi; Animal Models of Varicella Zoster Virus Infection. Pathogens 2013, 2, 364-382, 10.3390/pathogens2020364.
- Keith W Jarosinski; John E Carpenter; Erin M Buckingham; Wallen Jackson; Kevin Knudtson; Jennifer F Moffat; Hirohito Kita; Charles Grose; Cellular Stress Response to Varicella-Zoster Virus Infection of Human Skin Includes Highly Elevated Interleukin-6 Expression. Open Forum Infectious Diseases 2018, 5, ofy118, 10.1093/ofid/ofy118.

