Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ceyda Acilan | + 6372 word(s) | 6372 | 2022-01-25 07:34:57 | | | |
| 2 | Vivi Li | -7 word(s) | 6365 | 2022-02-08 09:46:51 | | | | |
| 3 | Vivi Li | Meta information modification | 6365 | 2022-02-10 11:25:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Acilan, C. Keep Calm and Carry on with Extra Centrosomes. Encyclopedia. Available online: https://encyclopedia.pub/entry/19199 (accessed on 24 June 2026).
Acilan C. Keep Calm and Carry on with Extra Centrosomes. Encyclopedia. Available at: https://encyclopedia.pub/entry/19199. Accessed June 24, 2026.
Acilan, Ceyda. "Keep Calm and Carry on with Extra Centrosomes" Encyclopedia, https://encyclopedia.pub/entry/19199 (accessed June 24, 2026).
Acilan, C. (2022, February 08). Keep Calm and Carry on with Extra Centrosomes. In Encyclopedia. https://encyclopedia.pub/entry/19199
Acilan, Ceyda. "Keep Calm and Carry on with Extra Centrosomes." Encyclopedia. Web. 08 February, 2022.
Copy Citation
Aberrations in the centrosome number and structure can readily be detected at all stages of tumor progression and are considered hallmarks of cancer. Centrosome anomalies are closely linked to chromosome instability and, therefore, are proposed to be one of the driving events of tumor formation and progression. This concept, first posited by Boveri over 100 years ago, has been an area of interest to cancer researchers.
cancer
centrosome
clustering
multipolar spindles
chromosomal instability
1. Introduction
Centrosomes are the major microtubule-organizing centers of the animal cell and are primarily known for their role in the regulation of microtubule nucleation [1]. Centrosomes also regulate other microtubule-dependent cell processes, such as cell shape and polarity in interphase cells [2]. Additionally, centrosomes function as a basal body for the primary cilium in quiescent cells [3].
Animal cells normally contain one or two centrosomes each, depending on their progression through the cell cycle. Mitosis generates two daughter cells, each possessing one centrosome inherited from the mother cell. A mature and functional centrosome is composed of two centrioles, a mother and a daughter, surrounded by pericentriolar material (PCM). During the S phase, centriole duplication is initiated on the wall of the mother centriole, while additional PCM components are trafficked to the centrosome via microtubules in a cytoplasmic dynein-dependent process [4], though some components can be recruited in a microtubule-independent manner [5]. This generates two centrosomes, which have a pair of centrioles surrounded by PCM. At the end of G2, phosphorylation of linker proteins that bridge between mother centrioles allows the centrosomes to be separated. Subsequently, the two centrosomes serve as the core of the mitotic spindle poles, guiding bipolar and symmetrical division to ensure even distribution of genetic material [6]. Disruptions in any one of these events may lead to centrosomal anomalies [7] that are associated with various physiological conditions or diseases, including reproductive disorders, ciliopathies, or cancer.
Due to a variety of factors, tumor cells frequently exhibit extra centrosomes (greater than two), a state often referred to as centrosome amplification (CA), which has been reported in a wide array of human cancers including solid tumors of various origins and hematological malignancies [8]. CA has long been considered a contributing factor to chromosomal instability (CIN) [9] and even proposed as a biomarker for personalized therapy for some types of cancers [10]. Frequently, extra centrosomes lead to the formation of multipolar spindles (MPS) during mitosis. As a consequence of a multipolar division, the daughter cells are likely not viable, since significant amounts of genetic material will fail to segregate properly and the cell will not inherit a full copy of the genome. Yet, cancer cells are able to circumvent MPS formation by clustering extra centrosomes in two poles, enabling bipolar division. Accumulated research has improved researchers' understanding that the centrosome-clustering mechanism is a key defense against cell death induced by multipolar division. Through clustering extra centrosomes, cancer cells can survive the potential consequences associated with CIN.
2. Control of Centrosome Biology
The centrosome duplication cycle is a heavily regulated intracellular process occurring during the cell cycle at roughly the same time as DNA replication. The regulation of the centrosome duplication cycle ensures that the centrosome is duplicated only once per cell cycle, in parallel to limiting DNA replication to a single occurrence per cell cycle. Coordination of these events is linked with the activity of cyclins and cyclin-dependent kinases (CDK) [11]. Protein levels of cyclins fluctuate throughout the cell cycle; for example, cyclin E levels peak at the G1-S transition [12], whereas expression of cyclin A and cyclin B is maximal during G2 and M phases [13]. When the intracellular levels of cyclins are sufficiently high, they activate their corresponding CDKs and form cyclin/CDK complexes. Given that the initiation of centrosome duplication occurs at late G1-S, it is not surprising to find out that the cyclin E/CDK2 complex regulates this event [11]. Both centrosome duplication and DNA replication are under the control of CDK2, which is activated by hyperphosphorylation of the retinoblastoma protein (pRB) [14]. As centrosome duplication is initiated, cyclin E/CDK2 phosphorylates nucleophosmin, inducing its dissociation from the centrosome. This process prevents premature centriole splitting, and a nonphosphorylatable form of nucleophosmin was demonstrated to be sufficient to prevent centrosome duplication [15]. As centrosome duplication progresses, in addition to the cyclin E/CDK2 complex, there is a requirement for activity and regulation by cyclin A/CDK2 [16]. Subsequently as centrosome duplication enters its later stages, dissolution of centriole linkers at G2/M transition is controlled by cyclin B/CDK1-mediated phosphorylation of Eg5 and Nek2-mediated phosphorylation of centriole linker proteins [17][18][19][20]. Further activity of cyclin/CDK complexes in this process can be seen because centrosome duplication in somatic animal cells requires the phosphorylation of pRB. Overexpression of E2F, a downstream effector of pRB, was found to be sufficient to override the effect caused by the expression of a non-phosphorylatable mutant of pRB, indicating E2F is the major effector of pRB pathway on centrosome duplication [16].
Duplication of centrosomes is also controlled intra-centrosomally. The Polo-like kinase PLK4 is known as the master regulator of centrosome duplication [21]. PLK4 is required for the biogenesis of a new pro-centriole [22]. In addition, this study also identified several other proteins necessary for procentriole formation: HsSAS-6, CPAP, CEP135, CP110, and γ-tubulin are required at different stages of procentriole formation [22]. Other studies have identified more key players: STIL contributes to centriole biogenesis via interplay with PLK4. PLK4 binds to and activates STIL by phosphorylation [23], and vice versa [24][25][26]. Importantly, the coiled-coil domain of STIL stabilizes PLK4, whereas the C-terminal domain of STIL removes excessive PLK4 activity, ensuring the formation of a single procentriole perpendicular to the wall of the mother centriole [27]. PLK4 activity is also regulated by auto-phosphorylation [28][29][30]. Independent studies have demonstrated that the interplay between PLK4 and STIL is the key to proper centriole biogenesis; this interaction is crucial for centriolar recruitment of HsSAS6 [26]. Notably, depletions of PLK4, STIL, or HsSAS-6 have been shown to block centrosome duplication [31][32][33]. Furthermore, CEP63 plays an early role in centriole biogenesis, preceding recruitment of HsSAS-6 [34]. Other CEP family proteins, CEP152 and CEP192, also interact with PLK4, and the conversion of PLK4 from a CEP192-tethered-and-sequestered state to a CEP152-bound-procentriole-assembly state is a critical requirement for centriole duplication [35][36].
Another feature of centrosome biology is centriole elongation (CE), which is initiated after procentriole formation. CE is regulated by several factors including CPAP, CP110, and CEP120, and is controlled by their activities and interactions. Centrosomal protein 4.1-associated protein (CPAP) is required to incorporate microtubules after pro-centriole assembly [37], and CP110 has been identified as a distal end-capping protein [38][39]. In addition, CEP120 interacts with CPAP and positively regulates CE [40]. A study using proteasome inhibitors demonstrated that inhibition of cellular proteolysis increases centriole length: observed centrioles were approximately four times normal length, showing that proper CE is dependent upon normal cellular proteolysis [41]. In this study, a siRNA screen uncovered several key centrosomal proteins. In addition to the aforementioned CPAP and CP110, seven other proteins were identified as CE regulators: FOP, CAP350, HsSAS-6, CEP170, ninein, C-Nap1, and CEP97 [41].
Since centrosome biology is regulated by many factors, dysregulation of any of them could cause centrosomal structural defects or aberrant centrosome numbers, leading to aneuploidy and chromosomal instability. Given that CA is a common feature of tumors, it is important to reveal the underlying mechanisms that cause CA.
3. Centrosome Amplification in Tumors and the Causes
Centrosome amplification in human cancers was first described by Boveri [42] more than a century ago and has gained attention over eighty years later when the discovery of the relation between the loss of p53 and centrosome amplification was made [43][44]. Subsequent studies added to this finding and researchers began to understand the causes of centrosome amplification in cancers better. Supernumerary centrosomes were observed in many clinical tumor samples [8], precancerous lesions [45], and cancer cell lines [46]; CA is also associated with poor patient prognosis [8]. Identification of the specific mechanisms of CA in cancers may help suppress cancer growth with inhibitors targeting CA mechanisms.
The question has always been whether CA was a cause or consequence of cancer, and some recent research has shown that under some specific conditions, amplification of centrosomes can promote tumorigenesis and tumor invasion [47]. One study has demonstrated that CA can initiate tumorigenesis in flies [48]. Similarly, a more recent study found that elevation of centrosome numbers increased tumor initiation in a mouse intestinal tumor model [49]. This study also showed the presence of supernumerary centrosomes was sufficient to drive aneuploidy and the development of spontaneous tumors in multiple tissues [49]. CA is also associated with increased invasiveness of tumors, and a study strikingly found that cells with extra centrosomes could induce invasion of neighboring cells in breast cancer via paracrine signaling, and secreted IL-8 was identified as a crucial factor for the induction of invasion via HER2 activation [50]. This study suggests that while only a small proportion of cells within a heterogeneous tumor mass may possess supernumerary centrosomes, these cells can exert a disproportionate effect upon tumor progression. Given that CA is increasingly associated with multiple aspects of tumor biology, it is important to understand the mechanisms that cause CA in tumors.
Mechanisms of centrosome amplification in cancers can be divided into two major classes: (i) overproduction of centrosomes due to the loss of tight control over the centrosome duplication process arising from pathway-inactivating genetic mutations and/or overexpression of centrosome duplication factors (direct centrosome amplification); and (ii) other cell biological events, which cause relative centrosome amplification, such as cell-cell fusions, failures of cytokinesis, premature centriole disengagement or centrosome (PCM) fragmentation (indirect centrosome amplification) (Summarized in Figure 1).
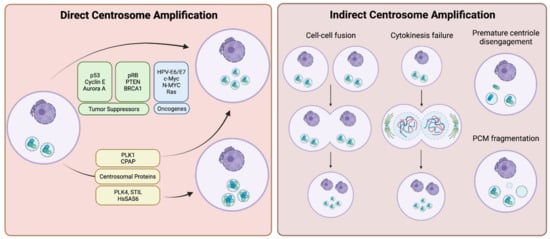
Figure 1. Centrosome amplification may occur directly through molecular changes within the cell such as overexpression of proteins regulating the centrosome cycle, presence of viral oncogenes, etc. (left panel), or indirectly via gross cellular anomalies, such as cellular fusion or premature centriole disengagement (right panel). Created with BioRender.com.
3.1. Direct Centrosome Amplification
The major route for the presence of supernumerary centrosomes in cancers is the deregulation of the centrosome duplication cycle. Many positive and negative regulators contribute to the control of the centrosome cycle, including tumor suppressors, such as p53, pRB, PTEN, or BRCA1. Loss of function mutations of these genes are associated with CA in various types of tumors. In addition, misexpression of proto-oncogenes, including E6, E7, Myc, and Ras is also associated with CA. Furthermore, changes in the genetic and epigenetic background in cancers can lead to the overexpression of centrosome duplication factors, such as PLK4, STIL, and HsSAS-6, which leads directly to CA by overproducing new centrioles.
3.1.1. Tumor Suppressors and Proto-Oncogenes
p53 is a well-defined tumor suppressor transcription factor that causes cell cycle arrest or apoptosis in response to DNA damage or other cellular stresses. In addition to being a tumor suppressor transcription factor, p53 also localizes to centrosomes, and the timing of the association of p53 with the centrosome is suggested to be an important regulatory step during mitosis [51][52]. Two main regulatory processes of centrosome duplication involve control by p53: initiation of centrosome duplication during late G1 [53], and suppression of a second duplication event during S and G2 [44]. Loss of p53 is one of the well-characterized causes of centrosome amplification in cancers [43]. Under p53−/− conditions, overexpression of p21Waf1/Cip1 was shown to partially revert the CA phenotype and reintroduction of wild type p53 completely reverted the p53−/− effects; suggesting p53 regulates centrosome duplication through multiple pathways, including a pathway where p21 functions as the effector protein [44][54]. Moreover, constitutive activation of the Cyclin E/CDK2 complex resulted in the uncoupling of centrosome duplication from DNA replication in MEFs derived from a p53 null mouse, and as a potent inhibitor of the Cyclin E/CDK2 complex [55], p21 was found to be pivotal in the coordination of these two key cell biological events [56]. Furthermore, both CDK2 and CDK4 have been identified as critical mediators of CA caused by loss of p53 [57]. Other proteins that are key players in cell cycle progression and inhibition are also critical to regulation of centrosome duplication; overexpression of the cell cycle inhibitory proteins, p16INK4A, p21Waf1 and p27Kip1, will block centrosome duplication [14][16][58]. Additionally, GADD45, a target of p53, has also been identified as a contributor to the regulation of centrosome duplication: GADD45−/− mice exhibit many of the characteristics observed in p53−/− mice including CA [58].
Whereas the loss or inactivation of p53 alone is sufficient to induce centrosome amplification in mouse cells, p53 silencing in human cells is not enough for the generation of CA, indicating additional factors are necessary. Numerous studies have given us clues about some of these factors that can work in conjunction with a downregulation of p53 functionality to drive CA in humans. One study identified cyclin E overexpression as a requirement to achieve CA by p53 inactivation in human bladder cancer cells [59]. Another demonstrated that overexpression of Aurora A kinase induces CA and aneuploidy in not only mouse NIH 3T3 cells, but also human breast cancer cells [60]. Given that Aurora A phosphorylates p53 at S315 and marks it for MDM2-dependent degradation [61], it is thought that Aurora A at least partially contributes to CA by driving p53 inactivation [62]. However, Aurora A overexpression, along with that of other mitotic kinases, can also contribute to both CA and polyploidy via another method: in a p53−/− background, overexpression of these kinases indirectly led to CA not through errors or deregulation of the centrosome duplication machinery, but instead by permitting cell viability to persist after defects in cytokinesis [63][64][65]. A more recent study also demonstrated that altered expression of NDRG1 (N-Myc down-regulated gene 1), a p53-inducible gene, affected centrosome number. NDRG1 was shown to directly interact with γ-tubulin and this interaction was reduced significantly in p53-knockout cells. This interaction and the function of NDRG1 was characterized as a pivotal component for normal centrosome homeostasis [66][67]. Taken together, many studies have shown the loss or inactivation of p53 causes centrosome amplification likely through multiple pathways, although the centrosomal targets and protein interactions of these processes still needs further research.
Another important tumor suppressor protein involved in CA is pRB. Much of researchers' understanding of how pRB contributes to CA has come from studies on human papilloma virus (HPV)-encoded oncoprotein E7 [68]. Expression of HPV-E7 destabilizes pRB and interferes with the CDK inhibitor p21 [69]. Interestingly, expression of E7 leads to abnormal centrosome synthesis occurring prior to nuclear abnormalities developing, whereas expression of the E6 oncoprotein leads to the aberrant centrosome duplication occurring at the same time as nuclear abnormalities [68]. Given that expression of E6 primarily targets p53 for ubiquitin-dependent degradation, it is speculated that E6 supports the survival of the cells with mitotic disorders [65]. It has also been shown that acute loss of pRB can cause CA and concomitant chromosomal instability in murine primary fibroblasts and a similar induction of CA and aneuploidy is observed when RNAi-mediated knockdown is performed in human primary fibroblasts [70].
Several other tumor suppressor genes have been demonstrated to be involved in CA when misregulated. BRCA1 is frequently mutated in cancers, and those mutations have been associated with CA [71]. BRCA1 upregulates p21 [72], suggesting that p21′s role in inducing CA may arise from the effects of BRCA1 mutations in addition to, or in place of, the aforementioned p53-p21 axis. Additionally, PTEN redistributes from nuclear and cytoplasmic localizations to the centrosomes as cells enter mitosis, peaking in prophase and prometaphase and this localization was characterized to protect the integrity of mitotic centrosomes [73]. Inhibition of Akt prevented the recruitment of PTEN to centrosomes, and reduction of PTEN and Akt levels resulted in centrosomal defects, suggesting both are required for the proper regulation of mitotic centrosomes [73].
Other than the loss of function mutations of tumor suppressor genes, oncogenic induction of tumor cells also results in amplified centrosomes. MYC and Ras are well-characterized oncogenes and both of them were shown to contribute to CA. Overexpression of c-MYC was found to induce CA by inhibiting the negative regulator of PLK4, SCF/CUL1, while promoting activity of the positive regulator, Cyclin E/CDK2 [74]. The oligopeptidase tripeptidyl peptidase II (TPPII) was found to be involved in CA, being upregulated in Burkitt lymphoma cells overexpressing c-MYC [75][76]. Overexpression of TPPII disrupted centrosomes via centriole multiplication, a state where multiple procentrioles are nucleated from a single mother centriole, and a subsequent knockdown and/or inactivation of TPPII inhibited c-MYC-induced centrosome errors. Thus, TPPII is a necessary factor for c-MYC induced CA in tumors [77]. In addition to c-MYC, N-MYC was also shown to contribute to CA in a p53-dependent manner [78]. This study found that in order for N-MYC directed CA to occur, the cell requires MDM2-mediated suppression of p53 activity; furthermore, reactivation of p53 with Nutlin-3A was sufficient to completely inhibit N-MYC directed CA [78]. Interestingly, a study examining the contribution of Ras and c-MYC to breast cancer tumorigenesis compared breast cancer initiation by c-Myc and K-Ras (G12D) in early malignancy in vivo, and intriguingly found that while both could lead to CA in tumors, only K-Ras could induce CA during pre-malignancy. In this case, CA was associated with increased expression of Cyclin D1, CDK4, and Nek2 [79]. This again demonstrates that there are many pathways that lead to CA. It also suggests that some are more likely to be exploited at particular stages of tumorigenesis than others.
3.1.2. Centrosomal Proteins
The Polo-like kinases are a family of five Ser/Thr kinases (PLK1-5) that have roles in cell cycle progression, the centrosome cycle, and mitosis. Centrosomal localization of all PLK isoforms have been reported in humans [80]. PLK4 is the keystone centrosomal duplication protein and increased expression of PLK4 has been reported in cancers originating from different tissues and organs including colon, stomach, breast, prostate, and brain [8][81][82][83][84][85]. By contrast, decreased PLK4 expression is observed in hepatocellular carcinoma [86] and hematological malignancies [87]. mRNA expression of PLK4 is regulated by p53 through recruitment of HDACs to the promoter region of the PLK4 [88] and loss of p53 in tumors is discussed as a contributing factor to the increased PLK4 expression [89]. Tumors that developed after PLK4 induced CA were also correlated with reduced p53 expression [49]. In addition, expression of the HPV-16 E7 oncoprotein was found to increase PLK4 levels [90]. Different studies in p53-deficient mice has shown that overexpression of PLK4 can accelerate tumorigenesis in epidermis [91] as well as induce CA, tissue hyperplasia, and loss of primary cilia [92].
PLK4 activity is crucial for centriole biogenesis and excess PLK4 levels lead to production of extra centrioles around a mother centriole, forming a structure named a rosette (or flower) centrosome due to its unique shape [22][93] (Figure 2). The first observation of rosette centrosomes was made in 1971 using thin-section electron microscopy [94][95]. Later, Habedanck et al. (2005) reported the formation of ‘‘flower-like structures’’ in cells overexpressing PLK4 [21]. Kleylein-Sohn et al. (2007) showed multiple centrioles in rosettes form during S phase and persist throughout S and G2 phases, suggesting rosette centrosomes are functional as a whole structure and cycle like normal centrosomes [22]. Kuriyama (2009) showed simultaneous overexpression of PLK4, HsSAS6, and SAS4 in CHO cells resulted in the formation of rosette centrosomes [95]. In addition, Cosenza et al. (2017) observed the presence of rosette centrosomes in primary tumor samples including multiple myeloma, glioblastoma and colon cancer samples, highlighting that the generation of these structures is observable in naturally occurring tumors, and are not just an artifact of genetic or pharmacological manipulation of cells [93]. The study also identified that overexpression of STIL is capable of generating rosette centrosomes [93]. A recent paper from Ching et al. showed that centrioles in olfactory sensory neurons are amplified in precursor cells via formation of centriole rosette structures, suggesting rosette centrosome formation is also required for the generation of normal multiciliated cells [96].
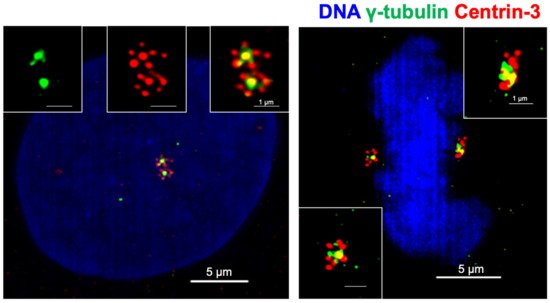
Figure 2. Centrioles in cancer cells form rosette like structures upon overexpression of PLK4. Rosette centrosomes can be readily seen in both interphase cells (left) and in mitosis (right). Blue: DNA (DAPI); green: centrosomes (γ-tubulin); red: centrioles (Centrin-3).
Inhibition of PLK4 with small molecule inhibitors has been considered as a therapeutic option in cancers [97][98]. A reversible PLK4 inhibitor, centrinone, led to centrosome depletion in cells, but was found to be effective at inhibiting proliferation only in normal cells; treatment resulted in a senescent-like state that was dependent upon p53 [99]. However, this study found that cancer cells were capable of proliferating even after centrosome loss induced by centrinone treatment and that after washout of the inhibitor, the cancer cells reverted to a distribution of centrosome number similar to untreated cells over time. This suggests that centrosome loss affects normal cells and cancer cells in different ways. A second inhibitor, CFI-400945, described as a potent and selective PLK4 inhibitor, was found effective in cancers, and taken into clinical trials [100][101]. However, subsequent studies have suggested that there may be important off-target effects of CFI-400945 to be considered [102][103] and the debate continues with the suggestion that CFI-400945 may promote overduplication of centrosomes at low concentrations, but deplete centrosomes at higher concentrations due to the stability and activity of PLK4 at those concentrations of treatment [104]. Nevertheless, PLK4 and PLK4 induced CA still provide interesting targets for small molecule inhibitors. A recent paper identified the ubiquitin ligase TRIM37 as responsible for cancer-specific vulnerability to PLK4 inhibition [105], and identification of vulnerability hot spots could help PLK4 targeting in cancers.
Other than PLK4, other PLK isoforms have also been investigated broadly. An increase in PLK1 expression is well documented in numerous cancers [106][107][108][109]. PLK1 was shown to regulate Mst2-Nek2A induced centrosome disjunction by phosphorylating Mst2 [20]. PLK1 also regulates separase activity and centriole disengagement [110]. In addition, depletion of PLK1 expression resulted in inhibition of CA in hydroxyurea-treated centrosome amplified U2OS cells, suggesting overexpression of PLK1 in cancer contributes to CA [111]. However, PLK1 overexpression has also been shown to result in segregational and cytokinetic defects, thus generating polyploid cells; this prevents the development of KRAS- and HER2-induced mammary tumors in vivo, suggesting the contribution of intracellular PLK1 expression levels to carcinogenesis could be both dose- and context-dependent [112].
PLK2 and PLK3 have been proposed to act as tumor suppressor proteins [86][113], and the loss of PLK2 was identified as a common change found in colorectal carcinomas, lending support to PLK2 being identified as a tumor suppressor [114]. Additionally, silencing of PLK2 in gastric cancer cells results in increased proliferation and decreased apoptosis [115]. Contrariwise, other research found that PLK2 activity promotes tumor growth and inhibits apoptosis of colorectal cancer cells in vitro and in vivo [116]. This research found that binding of PLK2 to the tumor suppressor Fbxw7 resulted in increased degradation of Fbxw7 and stabilization of Cyclin E; this suggested that PLK2 may act as a tumor supportive factor, may serve as a prognostic and diagnostic target and furthermore as a therapeutic target [116]. Similarly, PLK3 was defined as a tumor suppressor, and under hypoxic conditions, functions as a negative regulator of HIF-1α [117]. In addition, other studies have demonstrated that overexpression of PLK3 results in shortened survival time of patients with hepatocellular carcinoma [118][119], and PLK3 is upregulated in breast and ovarian cancers [120]. However, the PLK3 expression level appears to be downregulated in induced colon tumors [121], as well as in bladder [119], uterus [119], and head and neck tumors [122], suggesting that the tumor suppressor function for PLK3 cannot be generalized, but it depends on the context of the individual tumor type [123]. That the upregulation and downregulation of PLK1, PLK2, and PLK3 are capable of inducing tumorigenesis under different circumstances in different tumors underlies the multifaceted roles these kinases play in normal cell cycle progression, and how misexpression of these proteins can lead to tumor progression. These seemingly contradictory findings from different studies demonstrate that the functions of these PLK family proteins in cancer cell biology still need further investigation.
In addition to the PLK family, other proteins whose importance in centrosome regulation and contribution to CA have been partially elucidated. One such protein is HsSAS-6, whose expression level is important for the restriction of procentriole formation: overexpression of HsSAS-6 promotes the formation of the previously mentioned rosette structures consisting of excess procentrioles [124]. Increased HsSAS-6 mRNA and protein expression levels were observed in human primary colorectal carcinomas, and led to CA, mitotic abnormalities and increased chromosomal instability [125]. This finding led to an analysis of expression levels of HsSAS-6 in various cancer types that were catalogued in publicly available TCGA RNA-Seq databases; the analysis found that HsSAS-6 is overexpressed in numerous cancer tissues when compared to normal non-cancerous tissues; tissues determined to show overexpressed HsSAS-6 included bladder, kidney, breast, lung, prostate and pancreas [125]. Similarly, defects with procentriole assembly and subsequent chromosome segregation occur when CEP152 is defective: the mutations E21K and V8A have been identified to impair the interaction of CEP152 with PLK4 [36].
In addition to CA, the deregulation of centriole size was identified as another centrosomal feature of cancer cells. Recent research indicates that centriole over-elongation results in enlarged centrosomes and these have increased microtubule nucleation capacities and promote chromosome missegregation [46]. Overexpression of CPAP, an important CE factor, was shown to result in the formation of abnormally long centrioles. This led to cells having supernumerary MTOCs that then subsequently led to multipolar spindles and cytokinesis defects [126]. As a further consequence, centriole over-elongation also induces CA via ectopic procentriole formation [40][46][127].
3.2. Indirect Centrosome Amplification
The discussed alterations in gene expression can be considered direct mechanisms that result in CA, but supernumerary centrosomes can also originate from indirect mechanisms. These mechanisms include: (i) cell-cell fusion; (ii) failure of cytokinesis; (iii) premature centriole disengagement; and (iv) PCM fragmentation. These methods increase CA by means other than increased centriole biogenesis or duplication. (i) When cell-cell fusion occurs, two cells with the normal centrosome complement fuse together, generating a single cell that possesses twice the amount of DNA and double the number of centrosomes. (ii) A failure of cytokinesis means that a cell that has undergone both DNA replication and centrosome duplication but does not complete cytokinesis towards the end of mitosis, and reverts back to a single cell with twice the DNA and centrosome number present. (iii) Premature centriole disengagement drives early separation of the daughter centriole from the mother, and results in a cell with a normal DNA content, but more than two centrosomes. Of these excess centrosomes, at least two will consist of only a single centriole. (iv) PCM fragmentation occurs when significant amounts of PCM components aggregate in random places in the cytoplasm.
To identify the presence, prevalence, and impact of these types of CA, researchers have used fluorescent imaging. Antibodies specific to centrioles (e.g., Centrin-2, Centrin-3) and PCM components (e.g., γ-tubulin) in fixed cells, and fluorescently tagged proteins in live cells have proved invaluable tools in identifying each type of CA [127]. This approach of using both live and fixed cells is necessary; for example, it is infeasible to use fixed cells to understand the differences between cell-cell fusion and failures of cytokinesis and thus live cell imaging is critical.
3.2.1. Cell Fusion and Failures of Cytokinesis
Cell-cell fusions and cytokinesis failures result in cells that have twice the DNA and centrosome content. The fusion of two diploid (2n) cells generates a tetraploid (4n) hybrid cell, and often the nuclei of these fused cells remain separate from each other. However, sometimes the two nuclei will fuse and this can contribute to aneuploidy and cancer [128].
One key player in cell-cell fusion is RAD6. This protein, primarily known as an ubiquitin-conjugating enzyme, was found to be expressed and upregulated in metastatic breast tumors. The constitutive overexpression of RAD6 resulted in cell fusion, and subsequently, the generation of multinucleated cells, CA, multipolar mitotic spindles, and aneuploidy [129]. While this seems a straightforward association, cell fusion does not always cause aneuploidy and chromosomal instability, despite the fact that most of the fused cells have more than two centrosomes [130]. It is hypothesized that while CA caused by cell fusion can lead to multipolar spindle formation and therefore chromosomal instability in early stages of tumorigenesis, clonal outgrowth preferentially supports cells with a stable genome after tumor progression [128]. This could be achieved via centrosome clustering mechanisms, discussed in detail in later sections. Other than CA, cell fusion is also correlated with drug resistance and metastasis, serving a potential target for cancer therapy [131][132].
Cell division failure (failure of cytokinesis) can occur via many pathways. These pathways include the existence of a physical obstruction of the cleavage furrow, altered expression of regulators of cytokinesis, mutations in cytokinetic drivers, or mitotic slippage [133]. From these or other methods of failure of cytokinesis, the resulting cell will possess extra centrosomes as it re-enters interphase. As that cell progresses through the cell cycle, it will duplicate all of its centrosomes, ending with an excess number [134]. Failure of cytokinesis has significant tumorigenic potential: transient blockage of cytokinesis in p53−/− mouse mammary epithelial cells generates tetraploid cells, and these cells exhibited an increased frequency of chromosomal alterations and in vivo tumorigenic potential compared to their diploid counterparts [135]. It is postulated that tetraploid cells generated by cytokinesis defects are better able to tolerate the loss of chromosomes, thus allowing them to produce more viable aneuploid progeny [133][136]. Failure of cytokinesis was also reported to promote aneuploidy via multipolar mitosis in glioblastoma cells [137]. There are some reports that question a direct correlation between failure of cytokinesis and CA: one study indicated that repeated cleavage failure did not establish CA in untransformed human cells, and that cytokinetic failure resulted in only a small increase in CA in p53−/− HCT116 cells, but a relatively high increase in CHO cells. This suggests that the consequences of cleavage failure on increased CA frequency is likely cell type dependent [138].
A well-known and frequently observed characteristic of cancer cells is dicentric chromosomes. Chromosomes having two centromeres possess high potentiality to induce genome instability [139]. Centromeres of dicentric chromosomes tend to migrate towards the opposite poles during cell division, causing repetitive events of chromosomal breakages and re-ligations. Concomitant rearrangements of dicentric chromosomes occur due to the breakage-fusion-bridge (BFB) cycle [140][141]. Aberrations in gene copy numbers due to amplifications and deletions increases the likelihood of malignancy. Dicentric chromosomes are generated as a result of telomeric fusions [142] or form due to the presence of double strand breaks [143]. Repetitive DNA replications leads to telomere erosion and thus the formation of sticky ends on the chromosome. Chromosomal bridges generated by the fusions of telomeric regions of different chromosomes lead to cytokinetic failures, hence aneuploidy and cells bearing extra centrosomes [144][145][146]. Independent studies have discussed the correlation between centrosome amplification status and aneuploidy caused by telomere erosion. To clarify the origin of genome instability, researchers investigated centrosome amplification in different multiple myeloma stages, a cancer type well characterized by aneuploidy. They highlighted that centrosome amplification is frequently present even in early stages of myeloma. Moreover, they reported that there is no significant difference between ploidy subtypes (hyperdiploid: whole chromosome gains/losses; non-hyperdiploid: structural abnormalities and translocations) in terms of centrosome amplification [147]. This implied that telomere fusions and centrosome amplification contribute to CIN separately in the case of multiple myeloma. In another study, it was reported that telomere dysfunction and p16INK4a deficiency in breast cancer cooperatively caused centrosomal aberrations in both diploid and polyploid cells, even in the presence of functional p53 [148]. A novel molecular mechanism between telomeres and centrosomes was recently identified by another group. Telomerase transcriptional element interacting factor (TEIF), primarily known for its function in hTERT activation, was shown to localize to centrosomes in all stages of the cell cycle, and centrosomal recruitment was increased by telomere dysfunction [149]. In addition, EGF/PI3K signaling was identified as an important regulator of centrosomal recruitment of TEIF, resulting in centrosome amplification [150]. In colorectal cancers, a positive correlation between TEIF expression and centrosome amplification was reported [151], indicating that TEIF could be playing a crucial role mediating telomere dysfunction and centrosome aberrations.
3.2.2. Premature Centrosome Disengagement and PCM Fragmentation
While both processes involve the separation of parts of a centrosome, centriole disengagement is different from the centrosome separation. Centrosome separation occurs at the onset of mitosis as the duplicated centrosomes are separated to form the two spindle poles. As separation is initiated, centriolar linker proteins are phosphorylated, ensuring the connection between mother centrioles is severed by the onset of mitosis. This connection is made of linker proteins such as C-Nap1, rootletin, centlein, CEP68, and LRRC45 [152][153][154][155][156]. Initiation of separation occurs when Nek2A phosphorylates C-Nap1 and Rootletin [156][157][158][159][160] leading to an alteration of rootletin’s interaction with β-catenin [161]. After separation, centrosomes recruit additional PCM as they mature, thus allowing them to facilitate spindle assembly. Upon completion of mitosis, each daughter cell receives one centrosome, which consists of a mother-daughter pair that are tightly bound together. As the cell proceed through anaphase to next G1, the bound centriole pair disengage from each other, and after disengagement, the centrioles maintain connections to one another via the aforementioned centriolar linker proteins [18][162].
By comparison, premature centriole disengagement is not a normal phenomenon that occurs in healthy cells. When this process occurs, it will do so around metaphase and can result in the generation of multipolar spindles [127][163]. Centriole disengagement depends on the activity of separase, which is primarily known for its role in sister chromatid separation: separase hydrolyzes cohesin, which holds sister chromatids together [164]. However, separase also acts on centriole disengagement with numerous studies demonstrating an association between high separase activity and increased rates of multipolar spindle formation [165][166]. Separase functions by cleaving Scc1 (the cohesive keystone of the cohesin complex), and it has been observed that Scc1 is also localized to the centrosome, suggesting a role for Scc1 in maintaining centriole connections. Depletion of Scc1 also induced centriole splitting [167]. Furthermore, depletion of astrin, a microtubule and kinetochore protein, resulted in untimely separase activity and multipolar spindles, suggesting astrin contributes to the regulation of separase activity [166]. The Akt kinase interacting protein (Aki1) also seems to be involved in this process. Aki1 localizes to centrosomes and its depletion causes separase-dependent centriole splitting and multipolar spindles [167]. Separase’s ability to cleave both sister chromatids and centrioles was explored temporally with real-time imaging: using sensors, researchers could determine the point in mitosis at which separase’s activity was initiated. They found that centrosomal sensors were cleaved by separase before the onset of anaphase, and that this was earlier than they observed the chromosomal sensor being cleaved [110]. The activity of separase is inhibited by the spindle assembly checkpoint (SAC), thus mitotic delay or arrest may result in centriole disengagement. The SAC functions as a safety device to ensure proper, timely chromosome segregation and prevent missegregation [168]. SAC functions by delaying mitotic progression until the bipolar orientation of all chromatids has been achieved.
As SAC-induced mitotic delay persists, premature centriole disengagement and centrosome fragmentation follows; in order to maintain a bipolar spindle and division, the cell relies upon KIFC1(also known as HSET)-mediated centrosome clustering [163]. In addition, induction of mild replicative stress in non-cancerous cells was found to be sufficient to cause premature centriole disengagement, ultimately leading to transient multipolar spindles and lagging chromosomes. This early disengagement was related to the activity of CDK, PLK1, and ATR kinases during G2. This demonstrated that DNA damage can induce errors in centrosome cycle regulation [169].
Another mechanism by which premature centriole disengagement can occur was demonstrated by depleting the spindle-kinetochore proteins SKA3, CENP-E, or Cdc20. This led to the dysregulation of coordinated sister chromatid separation, a process termed cohesion fatigue; a consequence of cohesion fatigue was premature centriole disengagement and multipolar spindle formation [127][170]. Furthermore, recovery from pharmacological microtubule inhibition by colcemid or nocodazole leads to multipolar spindle formation with atypical centrioles at spindle poles. The poles formed here can be mono-centriolar or acentriolar in nature [128][159][160]. Additionally, nitrous oxide treatment [171] and heat shock [172] produced similar outcomes.
Independent from premature centriole disengagement, accumulating knowledge shows that PCM fragmentation is commonly observed in cancers. PCM fragmentation is defined as the generation of acentriolar PCM in random locations, from which microtubules can be nucleated and they are capable of functioning as spindle poles. One study examining CA in breast cancer found that high-grade tumors possess greater numbers of acentriolar centrosomes, suggesting PCM fragmentation may be common in breast cancers, and the rate of PCM fragmentation could increase with tumor stage and grade [173].
Research about the molecular mechanisms of PCM fragmentation has started to get attention, and several proteins have been identified as important contributing factors to centrosome integrity. Inactivation of Aurora A kinase, via either siRNA-mediated knockdown or a specific chemical inhibitor, is sufficient to cause PCM fragmentation and microtubule hyperstabilization [174]. Another key protein is kizuna, a centrosomal protein that is phosphorylated by PLK1 during mitosis and is critical for centrosome integrity and stabilization of the PCM [175][176]. Reduced expression of kizuna was shown to result in centrosome fragmentation and dispersion of PCM, leading to multipolarity and chromosome segregation errors [177]. This suggested that proper regulation and function of kizuna is a fundamental step of maintaining structural integrity of the centrosomes as they are exposed to the forces necessary to generate spindles [62]. In a recent study, a bioinformatic approach was used to identify novel mitotic components, and subsequent analysis revealed the role of chondrosarcoma-associated gene 1 protein (CSAG1) to be involved in maintenance of centrosome integrity during mitosis. Depletion of CSAG1 led to acentriolar multipolar spindle formation; this was especially pronounced in p53-compromised cells [178]. Moreover, siRNA-mediated knockdown of CEP164 was also demonstrated to result in the formation of multipolar spindles with acentriolar poles, identifying another protein that induces PCM fragmentation [179].
Pericentriolar satellites are granules that are found around the centrosome [180][181] and are involved in the recruitment of centrosomal components as well as modulation of microtubule organization [182]. Proper maintenance of these sites appears critical to preventing PCM fragmentation. Members of the CLASP (cytoplasmic linker-associated protein) family are known to be regulators of polarity in cell division, and alterations in CLASP expression leads to dysregulation of bipolar spindles [183]. CLASP1 regulates kinetochore-microtubule dynamics along with astrin and Kif2b [184]. However, a study found that siRNA-mediated knockdown of CLASP1/2 caused the generation of acentriolar spindle poles, suggesting the loss of these proteins could lead to PCM fragmentation. Further analysis revealed that CLASPs ensure spindle-pole integrity by recruiting ninein to pericentriolar satellites [185]. Additionally, CEP90 is characterized as a pericentriolar satellite localizing protein, and depletion of CEP90 causes spindle pole fragmentation and mitotic arrest, suggesting CEP90′s role at pericentriolar satellites is crucial for maintaining the integrity of spindle poles during mitosis [186]. Taken together, these studies demonstrate that pericentriolar satellites are vital for maintaining centrosomal integrity as additional forces are applied to the structures as the spindle is assembled.
References
- Bornens, M. The centrosome in cells and organisms. Science 2012, 335, 422–426.
- Godinho, S.A.; Kwon, M.; Pellman, D. Centrosomes and cancer: How cancer cells divide with too many centrosomes. Cancer Metastasis Rev. 2009, 28, 85–98.
- Sanchez, I.; Dynlacht, B.D. Cilium assembly and disassembly. Nat. Cell Biol. 2016, 18, 711–717.
- Balczon, R.; Varden, C.E.; Schroer, T.A. Role for microtubules in centrosome doubling in Chinese hamster ovary cells. Cell Motil. Cytoskelet. 1999, 42, 60–72.
- Hames, R.S.; Crookes, R.E.; Straatman, K.R.; Merdes, A.; Hayes, M.J.; Faragher, A.J.; Fry, A.M. Dynamic recruitment of Nek2 kinase to the centrosome involves microtubules, PCM-1, and localized proteasomal degradation. Mol. Biol. Cell 2005, 16, 1711–1724.
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624.
- Qi, F.; Zhou, J. Multifaceted roles of centrosomes in development, health, and disease. J. Mol. Cell Biol. 2021, 13, 611–621.
- Chan, J.Y. A clinical overview of centrosome amplification in human cancers. Int. J. Biol. Sci. 2011, 7, 1122–1144.
- Fukasawa, K. Centrosome amplification, chromosome instability and cancer development. Cancer Lett. 2005, 230, 6–19.
- Mahathre, M.M.; Rida, P.C.; Aneja, R. The more the messier: Centrosome amplification as a novel biomarker for personalized treatment of colorectal cancers. J. Biomed. Res. 2016, 30, 441–451.
- Lacey, K.R.; Jackson, P.K.; Stearns, T. Cyclin-dependent kinase control of centrosome duplication. Proc. Natl. Acad. Sci. USA 1999, 96, 2817–2822.
- Siu, K.T.; Rosner, M.R.; Minella, A.C. An integrated view of cyclin E function and regulation. Cell Cycle 2012, 11, 57–64.
- Yam, C.H.; Fung, T.K.; Poon, R.Y. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci. 2002, 59, 1317–1326.
- Matsumoto, Y.; Hayashi, K.; Nishida, E. Cyclin-dependent kinase 2 (Cdk2) is required for centrosome duplication in mammalian cells. Curr. Biol. 1999, 9, 429–432.
- Okuda, M.; Horn, H.F.; Tarapore, P.; Tokuyama, Y.; Smulian, A.G.; Chan, P.K.; Knudsen, E.S.; Hofmann, I.A.; Snyder, J.D.; Bove, K.E.; et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell 2000, 103, 127–140.
- Meraldi, P.; Lukas, J.; Fry, A.M.; Bartek, J.; Nigg, E.A. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat. Cell Biol. 1999, 1, 88–93.
- Blangy, A.; Lane, H.A.; d’Herin, P.; Harper, M.; Kress, M.; Nigg, E.A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 1995, 83, 1159–1169.
- Mardin, B.R.; Schiebel, E. Breaking the ties that bind: New advances in centrosome biology. J. Cell Biol. 2012, 197, 11–18.
- Fry, A.M.; Meraldi, P.; Nigg, E.A. A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 1998, 17, 470–481.
- Mardin, B.R.; Agircan, F.G.; Lange, C.; Schiebel, E. Plk1 controls the Nek2A-PP1gamma antagonism in centrosome disjunction. Curr. Biol. 2011, 21, 1145–1151.
- Habedanck, R.; Stierhof, Y.D.; Wilkinson, C.J.; Nigg, E.A. The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 2005, 7, 1140–1146.
- Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Habedanck, R.; Stierhof, Y.D.; Nigg, E.A. Plk4-induced centriole biogenesis in human cells. Dev. Cell 2007, 13, 190–202.
- Moyer, T.C.; Holland, A.J. PLK4 promotes centriole duplication by phosphorylating STIL to link the procentriole cartwheel to the microtubule wall. eLife 2019, 8, e46054.
- Moyer, T.C.; Clutario, K.M.; Lambrus, B.G.; Daggubati, V.; Holland, A.J. Binding of STIL to Plk4 activates kinase activity to promote centriole assembly. J. Cell Biol. 2015, 209, 863–878.
- Kratz, A.S.; Barenz, F.; Richter, K.T.; Hoffmann, I. Plk4-dependent phosphorylation of STIL is required for centriole duplication. Biol. Open 2015, 4, 370–377.
- Ohta, M.; Ashikawa, T.; Nozaki, Y.; Kozuka-Hata, H.; Goto, H.; Inagaki, M.; Oyama, M.; Kitagawa, D. Direct interaction of Plk4 with STIL ensures formation of a single procentriole per parental centriole. Nat. Commun. 2014, 5, 5267.
- Ohta, M.; Watanabe, K.; Ashikawa, T.; Nozaki, Y.; Yoshiba, S.; Kimura, A.; Kitagawa, D. Bimodal Binding of STIL to Plk4 Controls Proper Centriole Copy Number. Cell Rep. 2018, 23, 3160–3169.e4.
- Sillibourne, J.E.; Tack, F.; Vloemans, N.; Boeckx, A.; Thambirajah, S.; Bonnet, P.; Ramaekers, F.C.; Bornens, M.; Grand-Perret, T. Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 2010, 21, 547–561.
- Cunha-Ferreira, I.; Bento, I.; Pimenta-Marques, A.; Jana, S.C.; Lince-Faria, M.; Duarte, P.; Borrego-Pinto, J.; Gilberto, S.; Amado, T.; Brito, D.; et al. Regulation of autophosphorylation controls PLK4 self-destruction and centriole number. Curr. Biol. 2013, 23, 2245–2254.
- Guderian, G.; Westendorf, J.; Uldschmid, A.; Nigg, E.A. Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J. Cell Sci. 2010, 123, 2163–2169.
- Arquint, C.; Nigg, E.A. The PLK4-STIL-SAS-6 module at the core of centriole duplication. Biochem. Soc. Trans. 2016, 44, 1253–1263.
- Vulprecht, J.; David, A.; Tibelius, A.; Castiel, A.; Konotop, G.; Liu, F.; Bestvater, F.; Raab, M.S.; Zentgraf, H.; Izraeli, S.; et al. STIL is required for centriole duplication in human cells. J. Cell Sci. 2012, 125, 1353–1362.
- Arquint, C.; Sonnen, K.F.; Stierhof, Y.D.; Nigg, E.A. Cell-cycle-regulated expression of STIL controls centriole number in human cells. J. Cell Sci. 2012, 125, 1342–1352.
- Brown, N.J.; Marjanovic, M.; Luders, J.; Stracker, T.H.; Costanzo, V. Cep63 and cep152 cooperate to ensure centriole duplication. PLoS ONE 2013, 8, e69986.
- Hatch, E.M.; Kulukian, A.; Holland, A.J.; Cleveland, D.W.; Stearns, T. Cep152 interacts with Plk4 and is required for centriole duplication. J. Cell Biol. 2010, 191, 721–729.
- Park, S.Y.; Park, J.E.; Kim, T.S.; Kim, J.H.; Kwak, M.J.; Ku, B.; Tian, L.; Murugan, R.N.; Ahn, M.; Komiya, S.; et al. Molecular basis for unidirectional scaffold switching of human Plk4 in centriole biogenesis. Nat. Struct. Mol. Biol. 2014, 21, 696–703.
- Pelletier, L.; O’Toole, E.; Schwager, A.; Hyman, A.A.; Muller-Reichert, T. Centriole assembly in Caenorhabditis elegans. Nature 2006, 444, 619–623.
- Schmidt, T.I.; Kleylein-Sohn, J.; Westendorf, J.; Le Clech, M.; Lavoie, S.B.; Stierhof, Y.D.; Nigg, E.A. Control of centriole length by CPAP and CP110. Curr. Biol. 2009, 19, 1005–1011.
- Yadav, S.P.; Sharma, N.K.; Liu, C.; Dong, L.; Li, T.; Swaroop, A. Centrosomal protein CP110 controls maturation of the mother centriole during cilia biogenesis. Development 2016, 143, 1491–1501.
- Lin, Y.N.; Wu, C.T.; Lin, Y.C.; Hsu, W.B.; Tang, C.J.; Chang, C.W.; Tang, T.K. CEP120 interacts with CPAP and positively regulates centriole elongation. J. Cell Biol. 2013, 202, 211–219.
- Korzeniewski, N.; Cuevas, R.; Duensing, A.; Duensing, S. Daughter centriole elongation is controlled by proteolysis. Mol. Biol. Cell 2010, 21, 3942–3951.
- Boveri, T. Concerning the origin of malignant tumours by Theodor Boveri. Translated and annotated by Henry Harris. J. Cell Sci. 2008, 121 (Suppl. 1), 1–84.
- Fukasawa, K.; Choi, T.; Kuriyama, R.; Rulong, S.; Vande Woude, G.F. Abnormal centrosome amplification in the absence of p53. Science 1996, 271, 1744–1747.
- Tarapore, P.; Fukasawa, K. Loss of p53 and centrosome hyperamplification. Oncogene 2002, 21, 6234–6240.
- Pihan, G.A.; Wallace, J.; Zhou, Y.; Doxsey, S.J. Centrosome abnormalities and chromosome instability occur together in pre-invasive carcinomas. Cancer Res. 2003, 63, 1398–1404.
- Marteil, G.; Guerrero, A.; Vieira, A.F.; de Almeida, B.P.; Machado, P.; Mendonca, S.; Mesquita, M.; Villarreal, B.; Fonseca, I.; Francia, M.E.; et al. Over-elongation of centrioles in cancer promotes centriole amplification and chromosome missegregation. Nat. Commun. 2018, 9, 1258.
- Sabat-Pospiech, D.; Fabian-Kolpanowicz, K.; Prior, I.A.; Coulson, J.M.; Fielding, A.B. Targeting centrosome amplification, an Achilles’ heel of cancer. Biochem. Soc. Trans. 2019, 47, 1209–1222.
- Basto, R.; Brunk, K.; Vinadogrova, T.; Peel, N.; Franz, A.; Khodjakov, A.; Raff, J.W. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008, 133, 1032–1042.
- Levine, M.S.; Bakker, B.; Boeckx, B.; Moyett, J.; Lu, J.; Vitre, B.; Spierings, D.C.; Lansdorp, P.M.; Cleveland, D.W.; Lambrechts, D.; et al. Centrosome Amplification Is Sufficient to Promote Spontaneous Tumorigenesis in Mammals. Dev. Cell 2017, 40, 313–322.e5.
- Arnandis, T.; Monteiro, P.; Adams, S.D.; Bridgeman, V.L.; Rajeeve, V.; Gadaleta, E.; Marzec, J.; Chelala, C.; Malanchi, I.; Cutillas, P.R.; et al. Oxidative Stress in Cells with Extra Centrosomes Drives Non-Cell-Autonomous Invasion. Dev. Cell 2018, 47, 409–424.e9.
- Blair Zajdel, M.E.; Blair, G.E. The intracellular distribution of the transformation-associated protein p53 in adenovirus-transformed rodent cells. Oncogene 1988, 2, 579–584.
- Ciciarello, M.; Mangiacasale, R.; Casenghi, M.; Zaira Limongi, M.; D’Angelo, M.; Soddu, S.; Lavia, P.; Cundari, E. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J. Biol. Chem. 2001, 276, 19205–19213.
- Shinmura, K.; Bennett, R.A.; Tarapore, P.; Fukasawa, K. Direct evidence for the role of centrosomally localized p53 in the regulation of centrosome duplication. Oncogene 2007, 26, 2939–2944.
- Tarapore, P.; Horn, H.F.; Tokuyama, Y.; Fukasawa, K. Direct regulation of the centrosome duplication cycle by the p53-p21Waf1/Cip1 pathway. Oncogene 2001, 20, 3173–3184.
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512.
- Mussman, J.G.; Horn, H.F.; Carroll, P.E.; Okuda, M.; Tarapore, P.; Donehower, L.A.; Fukasawa, K. Synergistic induction of centrosome hyperamplification by loss of p53 and cyclin E overexpression. Oncogene 2000, 19, 1635–1646.
- Adon, A.M.; Zeng, X.; Harrison, M.K.; Sannem, S.; Kiyokawa, H.; Kaldis, P.; Saavedra, H.I. Cdk2 and Cdk4 regulate the centrosome cycle and are critical mediators of centrosome amplification in p53-null cells. Mol. Cell. Biol. 2010, 30, 694–710.
- Hollander, M.C.; Sheikh, M.S.; Bulavin, D.V.; Lundgren, K.; Augeri-Henmueller, L.; Shehee, R.; Molinaro, T.A.; Kim, K.E.; Tolosa, E.; Ashwell, J.D.; et al. Genomic instability in Gadd45a-deficient mice. Nat. Genet. 1999, 23, 176–184.
- Kawamura, K.; Izumi, H.; Ma, Z.; Ikeda, R.; Moriyama, M.; Tanaka, T.; Nojima, T.; Levin, L.S.; Fujikawa-Yamamoto, K.; Suzuki, K.; et al. Induction of centrosome amplification and chromosome instability in human bladder cancer cells by p53 mutation and cyclin E overexpression. Cancer Res. 2004, 64, 4800–4809.
- Zhou, H.; Kuang, J.; Zhong, L.; Kuo, W.L.; Gray, J.W.; Sahin, A.; Brinkley, B.R.; Sen, S. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat. Genet. 1998, 20, 189–193.
- Katayama, H.; Sasai, K.; Kawai, H.; Yuan, Z.M.; Bondaruk, J.; Suzuki, F.; Fujii, S.; Arlinghaus, R.B.; Czerniak, B.A.; Sen, S. Phosphorylation by aurora kinase A induces Mdm2-mediated destabilization and inhibition of p53. Nat. Genet. 2004, 36, 55–62.
- Fukasawa, K. Oncogenes and tumour suppressors take on centrosomes. Nat. Rev. Cancer 2007, 7, 911–924.
- Meraldi, P.; Honda, R.; Nigg, E.A. Aurora-A overexpression reveals tetraploidization as a major route to centrosome amplification in p53−/− cells. EMBO J. 2002, 21, 483–492.
- Borel, F.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Multiple centrosomes arise from tetraploidy checkpoint failure and mitotic centrosome clusters in p53 and RB pocket protein-compromised cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9819–9824.
- Nigg, E.A. Centrosome aberrations: Cause or consequence of cancer progression? Nat. Rev. Cancer 2002, 2, 815–825.
- Croessmann, S.; Wong, H.Y.; Zabransky, D.J.; Chu, D.; Mendonca, J.; Sharma, A.; Mohseni, M.; Rosen, D.M.; Scharpf, R.B.; Cidado, J.; et al. NDRG1 links p53 with proliferation-mediated centrosome homeostasis and genome stability. Proc. Natl. Acad. Sci. USA 2015, 112, 11583–11588.
- Kim, K.T.; Ongusaha, P.P.; Hong, Y.K.; Kurdistani, S.K.; Nakamura, M.; Lu, K.P.; Lee, S.W. Function of Drg1/Rit42 in p53-dependent mitotic spindle checkpoint. J. Biol. Chem. 2004, 279, 38597–38602.
- Duensing, S.; Duensing, A.; Crum, C.P.; Munger, K. Human papillomavirus type 16 E7 oncoprotein-induced abnormal centrosome synthesis is an early event in the evolving malignant phenotype. Cancer Res. 2001, 61, 2356–2360.
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997, 11, 2101–2111.
- Iovino, F.; Lentini, L.; Amato, A.; Di Leonardo, A. RB acute loss induces centrosome amplification and aneuploidy in murine primary fibroblasts. Mol. Cancer 2006, 5, 38.
- Xu, X.; Weaver, Z.; Linke, S.P.; Li, C.; Gotay, J.; Wang, X.W.; Harris, C.C.; Ried, T.; Deng, C.X. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol. Cell 1999, 3, 389–395.
- Somasundaram, K.; Zhang, H.; Zeng, Y.X.; Houvras, Y.; Peng, Y.; Zhang, H.; Wu, G.S.; Licht, J.D.; Weber, B.L.; El-Deiry, W.S. Arrest of the cell cycle by the tumour-suppressor BRCA1 requires the CDK-inhibitor p21WAF1/CiP1. Nature 1997, 389, 187–190.
- Leonard, M.K.; Hill, N.T.; Bubulya, P.A.; Kadakia, M.P. The PTEN-Akt pathway impacts the integrity and composition of mitotic centrosomes. Cell Cycle 2013, 12, 1406–1415.
- Korzeniewski, N.; Zheng, L.; Cuevas, R.; Parry, J.; Chatterjee, P.; Anderton, B.; Duensing, A.; Munger, K.; Duensing, S. Cullin 1 functions as a centrosomal suppressor of centriole multiplication by regulating polo-like kinase 4 protein levels. Cancer Res. 2009, 69, 6668–6675.
- Preta, G.; de Klark, R.; Gavioli, R.; Glas, R. The Enigma of Tripeptidyl-Peptidase II: Dual Roles in Housekeeping and Stress. J. Oncol. 2010, 2010, 128478.
- Stavropoulou, V.; Xie, J.; Henriksson, M.; Tomkinson, B.; Imreh, S.; Masucci, M.G. Mitotic infidelity and centrosome duplication errors in cells overexpressing tripeptidyl-peptidase II. Cancer Res. 2005, 65, 1361–1368.
- Duensing, S.; Darr, S.; Cuevas, R.; Melquiot, N.; Brickner, A.G.; Duensing, A.; Munger, K. Tripeptidyl Peptidase II Is Required for c-MYC-Induced Centriole Overduplication and a Novel Therapeutic Target in c-MYC-Associated Neoplasms. Genes Cancer 2010, 1, 883–892.
- Slack, A.D.; Chen, Z.; Ludwig, A.D.; Hicks, J.; Shohet, J.M. MYCN-directed centrosome amplification requires MDM2-mediated suppression of p53 activity in neuroblastoma cells. Cancer Res. 2007, 67, 2448–2455.
- Zeng, X.; Shaikh, F.Y.; Harrison, M.K.; Adon, A.M.; Trimboli, A.J.; Carroll, K.A.; Sharma, N.; Timmers, C.; Chodosh, L.A.; Leone, G.; et al. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene 2010, 29, 5103–5112.
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452.
- Liao, Z.; Zhang, H.; Fan, P.; Huang, Q.; Dong, K.; Qi, Y.; Song, J.; Chen, L.; Liang, H.; Chen, X.; et al. High PLK4 expression promotes tumor progression and induces epithelialmesenchymal transition by regulating the Wnt/betacatenin signaling pathway in colorectal cancer. Int. J. Oncol. 2019, 54, 479–490.
- Tian, X.; Zhou, D.; Chen, L.; Tian, Y.; Zhong, B.; Cao, Y.; Dong, Q.; Zhou, M.; Yan, J.; Wang, Y.; et al. Polo-like kinase 4 mediates epithelial-mesenchymal transition in neuroblastoma via PI3K/Akt signaling pathway. Cell Death Dis. 2018, 9, 54.
- Li, Z.; Dai, K.; Wang, C.; Song, Y.; Gu, F.; Liu, F.; Fu, L. Expression of Polo-Like Kinase 4(PLK4) in Breast Cancer and Its Response to Taxane-Based Neoadjuvant Chemotherapy. J. Cancer 2016, 7, 1125–1132.
- Korzeniewski, N.; Hohenfellner, M.; Duensing, S. CAND1 promotes PLK4-mediated centriole overduplication and is frequently disrupted in prostate cancer. Neoplasia 2012, 14, 799–806.
- Shinmura, K.; Kurabe, N.; Goto, M.; Yamada, H.; Natsume, H.; Konno, H.; Sugimura, H. PLK4 overexpression and its effect on centrosome regulation and chromosome stability in human gastric cancer. Mol. Biol. Rep. 2014, 41, 6635–6644.
- Pellegrino, R.; Calvisi, D.F.; Ladu, S.; Ehemann, V.; Staniscia, T.; Evert, M.; Dombrowski, F.; Schirmacher, P.; Longerich, T. Oncogenic and tumor suppressive roles of polo-like kinases in human hepatocellular carcinoma. Hepatology 2010, 51, 857–868.
- Ward, A.; Sivakumar, G.; Kanjeekal, S.; Hamm, C.; Labute, B.C.; Shum, D.; Hudson, J.W. The deregulated promoter methylation of the Polo-like kinases as a potential biomarker in hematological malignancies. Leuk. Lymphoma 2015, 56, 2123–2133.
- Li, J.; Tan, M.; Li, L.; Pamarthy, D.; Lawrence, T.S.; Sun, Y. SAK, a new polo-like kinase, is transcriptionally repressed by p53 and induces apoptosis upon RNAi silencing. Neoplasia 2005, 7, 312–323.
- Godinho, S.A.; Pellman, D. Causes and consequences of centrosome abnormalities in cancer. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369, 20130467.
- Korzeniewski, N.; Treat, B.; Duensing, S. The HPV-16 E7 oncoprotein induces centriole multiplication through deregulation of Polo-like kinase 4 expression. Mol. Cancer 2011, 10, 61.
- Sercin, O.; Larsimont, J.C.; Karambelas, A.E.; Marthiens, V.; Moers, V.; Boeckx, B.; Le Mercier, M.; Lambrechts, D.; Basto, R.; Blanpain, C. Transient PLK4 overexpression accelerates tumorigenesis in p53-deficient epidermis. Nat. Cell Biol. 2016, 18, 100–110.
- Coelho, P.A.; Bury, L.; Shahbazi, M.N.; Liakath-Ali, K.; Tate, P.H.; Wormald, S.; Hindley, C.J.; Huch, M.; Archer, J.; Skarnes, W.C.; et al. Over-expression of Plk4 induces centrosome amplification, loss of primary cilia and associated tissue hyperplasia in the mouse. Open Biol. 2015, 5, 150209.
- Cosenza, M.R.; Cazzola, A.; Rossberg, A.; Schieber, N.L.; Konotop, G.; Bausch, E.; Slynko, A.; Holland-Letz, T.; Raab, M.S.; Dubash, T.; et al. Asymmetric Centriole Numbers at Spindle Poles Cause Chromosome Missegregation in Cancer. Cell Rep. 2017, 20, 1906–1920.
- Dirksen, E.R. Centriole morphogenesis in developing ciliated epithelium of the mouse oviduct. J. Cell Biol. 1971, 51, 286–302.
- Kuriyama, R. Centriole assembly in CHO cells expressing Plk4/SAS6/SAS4 is similar to centriogenesis in ciliated epithelial cells. Cell Motil. Cytoskelet. 2009, 66, 588–596.
- Ching, K.; Stearns, T. Centrioles are amplified in cycling progenitors of olfactory sensory neurons. PLoS Biol. 2020, 18, e3000852.
- Holland, A.J.; Cleveland, D.W. Polo-like kinase 4 inhibition: A strategy for cancer therapy? Cancer Cell 2014, 26, 151–153.
- Zhao, Y.; Wang, X. PLK4: A promising target for cancer therapy. J. Cancer Res. Clin. Oncol. 2019, 145, 2413–2422.
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160.
- Mason, J.M.; Lin, D.C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176.
- Lohse, I.; Mason, J.; Cao, P.M.; Pintilie, M.; Bray, M.; Hedley, D.W. Activity of the novel polo-like kinase 4 inhibitor CFI-400945 in pancreatic cancer patient-derived xenografts. Oncotarget 2017, 8, 3064–3071.
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Polo-like kinase 4 inhibition produces polyploidy and apoptotic death of lung cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 1913–1918.
- Oegema, K.; Davis, R.L.; Lara-Gonzalez, P.; Desai, A.; Shiau, A.K. CFI-400945 is not a selective cellular PLK4 inhibitor. Proc. Natl. Acad. Sci. USA 2018, 115, E10808–E10809.
- Kawakami, M.; Mustachio, L.M.; Zheng, L.; Chen, Y.; Rodriguez-Canales, J.; Mino, B.; Kurie, J.M.; Roszik, J.; Villalobos, P.A.; Thu, K.L.; et al. Reply to Oegema et al.: CFI-400945 and Polo-like kinase 4 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E10810–E10811.
- Meitinger, F.; Ohta, M.; Lee, K.Y.; Watanabe, S.; Davis, R.L.; Anzola, J.V.; Kabeche, R.; Jenkins, D.A.; Shiau, A.K.; Desai, A.; et al. TRIM37 controls cancer-specific vulnerability to PLK4 inhibition. Nature 2020, 585, 440–446.
- Macmillan, J.C.; Hudson, J.W.; Bull, S.; Dennis, J.W.; Swallow, C.J. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann. Surg. Oncol. 2001, 8, 729–740.
- Wolf, G.; Elez, R.; Doermer, A.; Holtrich, U.; Ackermann, H.; Stutte, H.J.; Altmannsberger, H.M.; Rubsamen-Waigmann, H.; Strebhardt, K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997, 14, 543–549.
- Tokumitsu, Y.; Mori, M.; Tanaka, S.; Akazawa, K.; Nakano, S.; Niho, Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int. J. Oncol. 1999, 15, 687–692.
- Lee, S.Y.; Jang, C.; Lee, K.A. Polo-like kinases (plks), a key regulator of cell cycle and new potential target for cancer therapy. Dev. Reprod. 2014, 18, 65–71.
- Agircan, F.G.; Schiebel, E. Sensors at centrosomes reveal determinants of local separase activity. PLoS Genet. 2014, 10, e1004672.
- Liu, X.; Erikson, R.L. Activation of Cdc2/cyclin B and inhibition of centrosome amplification in cells depleted of Plk1 by siRNA. Proc. Natl. Acad. Sci. USA 2002, 99, 8672–8676.
- de Carcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montanes, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012.
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276.
- Matthew, E.M.; Yang, Z.; Peri, S.; Andrake, M.; Dunbrack, R.; Ross, E.; El-Deiry, W.S. Plk2 Loss Commonly Occurs in Colorectal Carcinomas but not Adenomas: Relationship to mTOR Signaling. Neoplasia 2018, 20, 244–255.
- Liu, L.Y.; Wang, W.; Zhao, L.Y.; Guo, B.; Yang, J.; Zhao, X.G.; Song, T.S.; Huang, C.; Xu, J.R. Silencing of polo-like kinase 2 increases cell proliferation and decreases apoptosis in SGC-7901 gastric cancer cells. Mol. Med. Rep. 2015, 11, 3033–3038.
- Ou, B.; Zhao, J.; Guan, S.; Wangpu, X.; Zhu, C.; Zong, Y.; Ma, J.; Sun, J.; Zheng, M.; Feng, H.; et al. Plk2 promotes tumor growth and inhibits apoptosis by targeting Fbxw7/Cyclin E in colorectal cancer. Cancer Lett. 2016, 380, 457–466.
- Yang, Y.; Bai, J.; Shen, R.; Brown, S.A.; Komissarova, E.; Huang, Y.; Jiang, N.; Alberts, G.F.; Costa, M.; Lu, L.; et al. Polo-like kinase 3 functions as a tumor suppressor and is a negative regulator of hypoxia-inducible factor-1 alpha under hypoxic conditions. Cancer Res. 2008, 68, 4077–4085.
- Weichert, W.; Denkert, C.; Schmidt, M.; Gekeler, V.; Wolf, G.; Kobel, M.; Dietel, M.; Hauptmann, S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br. J. Cancer 2004, 90, 815–821.
- Ando, K.; Ozaki, T.; Yamamoto, H.; Furuya, K.; Hosoda, M.; Hayashi, S.; Fukuzawa, M.; Nakagawara, A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J. Biol. Chem. 2004, 279, 25549–25561.
- Weichert, W.; Kristiansen, G.; Winzer, K.J.; Schmidt, M.; Gekeler, V.; Noske, A.; Muller, B.M.; Niesporek, S.; Dietel, M.; Denkert, C. Polo-like kinase isoforms in breast cancer: Expression patterns and prognostic implications. Virchows Arch. 2005, 446, 442–450.
- Dai, W.; Liu, T.; Wang, Q.; Rao, C.V.; Reddy, B.S. Down-regulation of PLK3 gene expression by types and amount of dietary fat in rat colon tumors. Int. J. Oncol. 2002, 20, 121–126.
- Dai, W.; Li, Y.; Ouyang, B.; Pan, H.; Reissmann, P.; Li, J.; Wiest, J.; Stambrook, P.; Gluckman, J.L.; Noffsinger, A.; et al. PRK, a cell cycle gene localized to 8p21, is downregulated in head and neck cancer. Genes Chromosomes Cancer 2000, 27, 332–336.
- Helmke, C.; Becker, S.; Strebhardt, K. The role of Plk3 in oncogenesis. Oncogene 2016, 35, 135–147.
- Strnad, P.; Leidel, S.; Vinogradova, T.; Euteneuer, U.; Khodjakov, A.; Gonczy, P. Regulated HsSAS-6 levels ensure formation of a single procentriole per centriole during the centrosome duplication cycle. Dev. Cell 2007, 13, 203–213.
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Nagura, K.; Kamo, T.; Okubo, Y.; Inoue, Y.; Kurabe, N.; Du, C.; Iwaizumi, M.; et al. SASS6 overexpression is associated with mitotic chromosomal abnormalities and a poor prognosis in patients with colorectal cancer. Oncol. Rep. 2015, 34, 727–738.
- Kohlmaier, G.; Loncarek, J.; Meng, X.; McEwen, B.F.; Mogensen, M.M.; Spektor, A.; Dynlacht, B.D.; Khodjakov, A.; Gonczy, P. Overly long centrioles and defective cell division upon excess of the SAS-4-related protein CPAP. Curr. Biol. 2009, 19, 1012–1018.
- Maiato, H.; Logarinho, E. Mitotic spindle multipolarity without centrosome amplification. Nat. Cell Biol. 2014, 16, 386–394.
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539.
- Shekhar, M.P.; Lyakhovich, A.; Visscher, D.W.; Heng, H.; Kondrat, N. Rad6 overexpression induces multinucleation, centrosome amplification, abnormal mitosis, aneuploidy, and transformation. Cancer Res. 2002, 62, 2115–2124.
- Lu, X.; Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. USA 2009, 106, 9385–9390.
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132.
- Fernandes, C.; Prabhu, P.; Juvale, K.; Suares, D.; Yc, M. Cancer cell fusion: A potential target to tackle drug-resistant and metastatic cancer cells. Drug Discov. Today 2019, 24, 1836–1844.
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45.
- Millband, D.N.; Campbell, L.; Hardwick, K.G. The awesome power of multiple model systems: Interpreting the complex nature of spindle checkpoint signaling. Trends Cell Biol. 2002, 12, 205–209.
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047.
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54.
- Telentschak, S.; Soliwoda, M.; Nohroudi, K.; Addicks, K.; Klinz, F.J. Cytokinesis failure and successful multipolar mitoses drive aneuploidy in glioblastoma cells. Oncol. Rep. 2015, 33, 2001–2008.
- Krzywicka-Racka, A.; Sluder, G. Repeated cleavage failure does not establish centrosome amplification in untransformed human cells. J. Cell Biol. 2011, 194, 199–207.
- Shen, Z. Genomic instability and cancer: An introduction. J. Mol. Cell Biol. 2011, 3, 1–3.
- McClintock, B. The behavior in successive nuclear divisions of a chromosome broken at meiosis. Proc. Natl. Acad. Sci. USA 1939, 25, 405–416.
- MacKinnon, R.N.; Campbell, L.J. The role of dicentric chromosome formation and secondary centromere deletion in the evolution of myeloid malignancy. Genet. Res. Int. 2011, 2011, 643628.
- Capper, R.; Britt-Compton, B.; Tankimanova, M.; Rowson, J.; Letsolo, B.; Man, S.; Haughton, M.; Baird, D.M. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007, 21, 2495–2508.
- Acilan, C.; Potter, D.M.; Saunders, W.S. DNA repair pathways involved in anaphase bridge formation. Genes Chromosomes Cancer 2007, 46, 522–531.
- Gisselsson, D.; Lv, M.; Tsao, S.W.; Man, C.; Jin, C.; Höglund, M.; Kwong, Y.L.; Jin, Y. Telomere-mediated mitotic disturbances in immortalized ovarian epithelial cells reproduce chromosomal losses and breakpoints from ovarian carcinoma. Genes Chromosomes Cancer 2005, 42, 22–33.
- Gisselsson, D.; Jonson, T.; Yu, C.; Martins, C.; Mandahl, N.; Wiegant, J.; Jin, Y.; Mertens, F.; Jin, C. Centrosomal abnormalities, multipolar mitoses, and chromosomal instability in head and neck tumours with dysfunctional telomeres. Br. J. Cancer 2002, 87, 202–207.
- Gisselsson, D.; Gorunova, L.; Höglund, M.; Mandahl, N.; Elfving, P. Telomere shortening and mitotic dysfunction generate cytogenetic heterogeneity in a subgroup of renal cell carcinomas. Br. J. Cancer 2004, 91, 327–332.
- Chng, W.J.; Ahmann, G.J.; Henderson, K.; Santana-Davila, R.; Greipp, P.R.; Gertz, M.A.; Lacy, M.Q.; Dispenzieri, A.; Kumar, S.; Rajkumar, S.V.; et al. Clinical implication of centrosome amplification in plasma cell neoplasm. Blood 2006, 107, 3669–3675.
- Domínguez, D.; Feijoo, P.; Bernal, A.; Ercilla, A.; Agell, N.; Genescà, A.; Tusell, L. Centrosome aberrations in human mammary epithelial cells driven by cooperative interactions between p16INK4a deficiency and telomere-dependent genotoxic stress. Oncotarget 2015, 6, 28238–28256.
- Gong, Y.; Sun, Y.; McNutt, M.A.; Sun, Q.; Hou, L.; Liu, H.; Shen, Q.; Ling, Y.; Chi, Y.; Zhang, B. Localization of TEIF in the centrosome and its functional association with centrosome amplification in DNA damage, telomere dysfunction and human cancers. Oncogene 2009, 28, 1549–1560.
- Zhao, J.; Zou, Y.; Liu, H.; Wang, H.; Zhang, H.; Hou, W.; Li, X.; Jia, X.; Zhang, J.; Hou, L.; et al. TEIF associated centrosome activity is regulated by EGF/PI3K/Akt signaling. Biochim. Biophys. Acta 2014, 1843, 1851–1864.
- Gao, Y.; Zhang, B. Expression of TEIF protein in colorectal tumors and its correlation with centrosome abnormality. Chin. J. Cancer 2009, 28, 1277–1282.
- Vlijm, R.; Li, X.; Panic, M.; Ruthnick, D.; Hata, S.; Herrmannsdorfer, F.; Kuner, T.; Heilemann, M.; Engelhardt, J.; Hell, S.W.; et al. STED nanoscopy of the centrosome linker reveals a CEP68-organized, periodic rootletin network anchored to a C-Nap1 ring at centrioles. Proc. Natl. Acad. Sci. USA 2018, 115, E2246–E2253.
- Bahe, S.; Stierhof, Y.D.; Wilkinson, C.J.; Leiss, F.; Nigg, E.A. Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 2005, 171, 27–33.
- He, R.; Huang, N.; Bao, Y.; Zhou, H.; Teng, J.; Chen, J. LRRC45 is a centrosome linker component required for centrosome cohesion. Cell Rep. 2013, 4, 1100–1107.
- Fang, G.; Zhang, D.; Yin, H.; Zheng, L.; Bi, X.; Yuan, L. Centlein mediates an interaction between C-Nap1 and Cep68 to maintain centrosome cohesion. J. Cell Sci. 2014, 127, 1631–1639.
- Yang, J.; Adamian, M.; Li, T. Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol. Biol. Cell 2006, 17, 1033–1040.
- Fry, A.M.; Mayor, T.; Meraldi, P.; Stierhof, Y.D.; Tanaka, K.; Nigg, E.A. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol. 1998, 141, 1563–1574.
- Mayor, T.; Hacker, U.; Stierhof, Y.D.; Nigg, E.A. The mechanism regulating the dissociation of the centrosomal protein C-Nap1 from mitotic spindle poles. J. Cell Sci. 2002, 115, 3275–3284.
- Mayor, T.; Stierhof, Y.D.; Tanaka, K.; Fry, A.M.; Nigg, E.A. The centrosomal protein C-Nap1 is required for cell cycle-regulated centrosome cohesion. J. Cell Biol. 2000, 151, 837–846.
- Hayward, D.G.; Fry, A.M. Nek2 kinase in chromosome instability and cancer. Cancer Lett. 2006, 237, 155–166.
- Bahmanyar, S.; Kaplan, D.D.; Deluca, J.G.; Giddings, T.H., Jr.; O’Toole, E.T.; Winey, M.; Salmon, E.D.; Casey, P.J.; Nelson, W.J.; Barth, A.I. beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008, 22, 91–105.
- Tsou, M.F.; Stearns, T. Controlling centrosome number: Licenses and blocks. Curr. Opin. Cell Biol. 2006, 18, 74–78.
- Karki, M.; Keyhaninejad, N.; Shuster, C.B. Precocious centriole disengagement and centrosome fragmentation induced by mitotic delay. Nat. Commun. 2017, 8, 15803.
- Wirth, K.G.; Wutz, G.; Kudo, N.R.; Desdouets, C.; Zetterberg, A.; Taghybeeglu, S.; Seznec, J.; Ducos, G.M.; Ricci, R.; Firnberg, N.; et al. Separase: A universal trigger for sister chromatid disjunction but not chromosome cycle progression. J. Cell Biol. 2006, 172, 847–860.
- Tsou, M.F.; Stearns, T. Mechanism limiting centrosome duplication to once per cell cycle. Nature 2006, 442, 947–951.
- Thein, K.H.; Kleylein-Sohn, J.; Nigg, E.A.; Gruneberg, U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J. Cell Biol. 2007, 178, 345–354.
- Nakamura, A.; Arai, H.; Fujita, N. Centrosomal Aki1 and cohesin function in separase-regulated centriole disengagement. J. Cell Biol. 2009, 187, 607–614.
- Musacchio, A.; Salmon, E.D. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 2007, 8, 379–393.
- Wilhelm, T.; Olziersky, A.M.; Harry, D.; De Sousa, F.; Vassal, H.; Eskat, A.; Meraldi, P. Mild replication stress causes chromosome mis-segregation via premature centriole disengagement. Nat. Commun. 2019, 10, 3585.
- Daum, J.R.; Potapova, T.A.; Sivakumar, S.; Daniel, J.J.; Flynn, J.N.; Rankin, S.; Gorbsky, G.J. Cohesion fatigue induces chromatid separation in cells delayed at metaphase. Curr. Biol. 2011, 21, 1018–1024.
- Brinkley, B.R.; Rao, P.N. Nitrous oxide: Effects on the mitotic apparatus and chromosome movement in HeLa cells. J. Cell Biol. 1973, 58, 96–106.
- Vidair, C.A.; Doxsey, S.J.; Dewey, W.C. Heat shock alters centrosome organization leading to mitotic dysfunction and cell death. J. Cell Physiol. 1993, 154, 443–455.
- Denu, R.A.; Zasadil, L.M.; Kanugh, C.; Laffin, J.; Weaver, B.A.; Burkard, M.E. Centrosome amplification induces high grade features and is prognostic of worse outcomes in breast cancer. BMC Cancer 2016, 16, 47.
- Asteriti, I.A.; Giubettini, M.; Lavia, P.; Guarguaglini, G. Aurora-A inactivation causes mitotic spindle pole fragmentation by unbalancing microtubule-generated forces. Mol. Cancer 2011, 10, 131.
- Cizmecioglu, O.; Hoffmann, I. Kizuna takes pole position. Nat. Cell Biol. 2006, 8, 1050–1051.
- Fry, A.M.; Baxter, J.E. Sealed with a Kiz: How Plk1 ensures spindle pole integrity. Dev. Cell 2006, 11, 431–432.
- Oshimori, N.; Ohsugi, M.; Yamamoto, T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat. Cell Biol. 2006, 8, 1095–1101.
- Sapkota, H.; Wren, J.D.; Gorbsky, G.J. CSAG1 maintains the integrity of the mitotic centrosome in cells with defective p53. J. Cell Sci. 2020, 133, jcs239723.
- Leber, B.; Maier, B.; Fuchs, F.; Chi, J.; Riffel, P.; Anderhub, S.; Wagner, L.; Ho, A.D.; Salisbury, J.L.; Boutros, M.; et al. Proteins required for centrosome clustering in cancer cells. Sci. Transl. Med. 2010, 2, 33ra38.
- Kubo, A.; Sasaki, H.; Yuba-Kubo, A.; Tsukita, S.; Shiina, N. Centriolar satellites: Molecular characterization, ATP-dependent movement toward centrioles and possible involvement in ciliogenesis. J. Cell Biol. 1999, 147, 969–980.
- Kubo, A.; Tsukita, S. Non-membranous granular organelle consisting of PCM-1: Subcellular distribution and cell-cycle-dependent assembly/disassembly. J. Cell Sci. 2003, 116, 919–928.
- Dammermann, A.; Merdes, A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J. Cell Biol. 2002, 159, 255–266.
- Mimori-Kiyosue, Y.; Grigoriev, I.; Sasaki, H.; Matsui, C.; Akhmanova, A.; Tsukita, S.; Vorobjev, I. Mammalian CLASPs are required for mitotic spindle organization and kinetochore alignment. Genes Cells 2006, 11, 845–857.
- Manning, A.L.; Bakhoum, S.F.; Maffini, S.; Correia-Melo, C.; Maiato, H.; Compton, D.A. CLASP1, astrin and Kif2b form a molecular switch that regulates kinetochore-microtubule dynamics to promote mitotic progression and fidelity. EMBO J. 2010, 29, 3531–3543.
- Logarinho, E.; Maffini, S.; Barisic, M.; Marques, A.; Toso, A.; Meraldi, P.; Maiato, H. CLASPs prevent irreversible multipolarity by ensuring spindle-pole resistance to traction forces during chromosome alignment. Nat. Cell Biol. 2012, 14, 295–303.
- Kim, K.; Rhee, K. The pericentriolar satellite protein CEP90 is crucial for integrity of the mitotic spindle pole. J. Cell Sci. 2011, 124, 338–347.
More
Information
Subjects:
Cell Biology; Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
3 times
(View History)
Update Date:
10 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No