Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yen-Lin Liu | + 1173 word(s) | 1173 | 2022-01-29 05:20:25 | | | |
| 2 | Beatrix Zheng | + 220 word(s) | 1393 | 2022-02-04 16:20:26 | | | | |
| 3 | Beatrix Zheng | + 220 word(s) | 1393 | 2022-02-04 16:21:04 | | | | |
| 4 | Beatrix Zheng | Meta information modification | 1393 | 2022-02-07 04:30:05 | | | | |
| 5 | Beatrix Zheng | Meta information modification | 1393 | 2022-02-08 12:20:28 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Liu, Y.; Chun-Yen, Y. Atypical Teratoid/Rhabdoid Tumor in Taiwan. Encyclopedia. Available online: https://encyclopedia.pub/entry/19095 (accessed on 24 June 2026).
Liu Y, Chun-Yen Y. Atypical Teratoid/Rhabdoid Tumor in Taiwan. Encyclopedia. Available at: https://encyclopedia.pub/entry/19095. Accessed June 24, 2026.
Liu, Yen-Lin, Yu Chun-Yen. "Atypical Teratoid/Rhabdoid Tumor in Taiwan" Encyclopedia, https://encyclopedia.pub/entry/19095 (accessed June 24, 2026).
Liu, Y., & Chun-Yen, Y. (2022, February 03). Atypical Teratoid/Rhabdoid Tumor in Taiwan. In Encyclopedia. https://encyclopedia.pub/entry/19095
Liu, Yen-Lin and Yu Chun-Yen. "Atypical Teratoid/Rhabdoid Tumor in Taiwan." Encyclopedia. Web. 03 February, 2022.
Copy Citation
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare, highly aggressive embryonal brain tumor most commonly presenting in young children. Older age, supratentorial site, and treatment with radiotherapy, chemotherapy, or both were significantly associated with better survival of patients with AT/RT in Taiwan.
atypical teratoid/rhabdoid tumor
CNS tumors
pediatric cancer
survival outcome
1. Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare and highly malignant cancer of the central nervous system (CNS). AT/RT represents 1 to 2% of all pediatric CNS tumors [1][2][3][4] and is the most common CNS malignant tumor in children under 3 years of age [1][5]. In children under the age of 1, AT/RT accounts for 40 to 50% of CNS malignancies [2]. It is more prevalent in males and in children of European descent [6][7]. AT/RT is characterized by loss-of-function alterations in the SMARCB1 gene on chromosome 22q11.2 in more than 95% of patients, with the remainder having mutations in SMARCA4, located on chromosome 19p13.2 [2][8][9][10]. AT/RTs have been found throughout the CNS, most commonly in the infratentorial region; their location may vary with age [2][11].
Pathologically, AT/RTs are embryonal tumors that have a rhabdoid morphology, as well as areas with primitive neuroectodermal, mesenchymal, and epithelial features [5]. Radiographically, AT/RT typically presents as a large, heterogeneous mass with varying degrees of necrosis, hemorrhage, and peritumoral edema, mostly within the CNS but sometimes along the cranial nerves or at the skull base [2].
AT/RTs are highly malignant in nature and are classified as Grade IV CNS tumors according to the World Health Organization (WHO) classification [12]. Even with intensive multimodality therapies, the prognosis of AT/RT is poor, with a 15–53% of survival rate at three years and a median survival of approximately 1 year [3][8][13][14]. Due to the rare occurrence of AT/RT, the optimal treatment has yet to be determined, and therapeutic approaches vary from institution to institution [1][13]. AT/RTs are most commonly managed using a multimodality treatment that includes surgery followed by chemotherapy, radiotherapy, high-dose chemotherapy with stem cell therapy (SCT), and intrathecal or intraventricular (IT/IVent) chemotherapy [2]. Although the extent of surgical resection has been proven to be associated with better outcomes, there is no universally accepted chemotherapy or radiotherapy regimen for AT/RT. Previous studies suggest that patients may have a longer disease-free survival with SCT [5]. To reduce the risk of neurocognitive toxicity in younger patients, radiotherapy may be delayed; however, this may affect the overall survival [2][15].
2. Current Insights
2.1. Demographic and Clinical Characteristics
The demographic and clinical characteristics of the study population are shown in Table 1. Of 47 enrolled patients with AT/RT, 29 were male (61.70%). The mean age of the patients was 66.87 (±109.32) months; 11 patients were younger than 12 months of age (23.40%), 15 were 12 to 35 months old (31.91%), and 21 were 36 months of age or older (44.68%). Regarding the tumor site, 46.81% of the tumors were in the infratentorial region or in the spine (n = 22), 29.79% were at an unspecified site (n = 14), and 23.40% were in the supratentorial region (n = 11). In this group, 24 patients received combined radiotherapy and chemotherapy (51.06%), 12 patients received chemotherapy only (25.53%), 6 patients received surgery only or no treatment (12.77%), and 5 patients received radiotherapy only (10.64%). In addition, 24 patients were diagnosed between 1999 and 2007 (51.06%), and 23 patients were diagnosed between 2008 and 2014 (48.94%). Nearly half of the patients (n = 21, 44.68%) were from Northern Taiwan, followed by Central Taiwan (n = 15, 31.91%) and then Southern Taiwan (n = 11, 23.40%).
Table 1. Demographic and clinical characteristics of the patients.
| Variables | Total (N = 47) * |
|---|---|
| Mean age at diagnosis (months) * Median age at diagnosis (months) |
66.87 (±109.32) 23.3 (12.5–87.9) |
| Age group (months) | |
| 0–11 months | 11 (23.40%) |
| 12–35 months | 15 (31.91%) |
| ≥36 months | 21 (44.68%) |
| Gender, Male | 29 (61.70%) |
| Residence Location | |
| Northern Taiwan | 21 (44.68%) |
| Central Taiwan | 15 (31.91%) |
| Southern Taiwan | 11 (23.40%) |
| Tumor site | |
| Supratentorial | 11 (23.40%) |
| Infratentorial or Spine | 22 (46.81%) |
| Unspecified nervous system or Others | 14 (29.79%) |
| Treatment | |
| Surgery or no treatment | 6 (12.77%) |
| Chemotherapy (including SCT) | 12 (25.53%) |
| RT | 5 (10.64%) |
| RT + CT | 24 (51.06%) |
| Diagnosis year | |
| 2008–2014 | 23 (48.94%) |
| 1999–2007 | 24 (51.06%) |
* Variables are expressed as mean ± standard deviation (SD) or median (interquartile range (IQR)) for continuous data, and n (%) for categorical data.
2.2. Distribution of Tumor Site and Treatment across Age Groups
In Table 2, which compares the tumor site and treatment across age groups, we noted a trend of a higher prevalence of infratentorial/spinal tumors in younger patients (n = 15) and of supratentorial tumors in older patients (n = 8) (p = 0.082). Children younger than 3 years of age more commonly had surgery only or no treatment (n = 6 and 0), and fewer received treatment with chemotherapy and/or radiotherapy than children 3 years or older (n = 20 and 21) (p = 0.026).
Table 2. Comparison of tumor site and treatment types for different age groups.
| Tumor Site | Age < 3 Years (N = 26) |
Age ≥ 3 Years (N = 21) |
Total (N = 47) |
p Value |
|---|---|---|---|---|
| Supratentorial | 3 (27%) | 8 (73%) | 11 | 0.082 a |
| Infratentorial or Spine | 15 (68%) | 7 (32) | 22 | |
| Unspecified nervous system or Others | 8 (57%) | 6 (43%) | 14 | |
| Treatment | (N = 26) | (N = 21) | (N = 47) | |
| Chemotherapy and/or radiotherapy | 20 (49%) | 21 (51%) | 41 | 0.026 b |
| Surgery or No treatment | 6 (100%) | 0 (0%) | 6 |
a Chi-square test of independence; b Fisher’s exact test.
We found no significant relationship between tumor site and treatment (Table 3; p = 0.6588).
Table 3. Comparison of tumor site for different treatment types.
| Tumor Site | Chemotherapy Only (N = 12) |
Radiotherapy with/ without Chemotherapy (N = 29) |
Surgery or No Treatment (N = 6) |
Total (N = 47) |
p Value * | |
|---|---|---|---|---|---|---|
| Infratentorial or Spine | 7 (32%) | 12 (54%) | 3 (14%) | 22 | 0.698 | |
| Supratentorial, Unspecified or Others | 5 (20%) | 17 (68%) | 3 (12%) | 25 |
* Fisher’s exact test.
2.3. Prognostic Factors
Kaplan–Meier analysis (Figure 1) showed that the survival probabilities of the patients who were aged ≥36 months (Figure 1a), whose tumor was located at a supratentorial site (Figure 1d), and who received radiotherapy (Figure 1e), were significantly higher (all p < 0.05 by log-rank test) than those of the other patients. Gender, residence location in Taiwan, and diagnosis year had no significant influence on survival (Figure 1b,c,f; all p > 0.05 by log-rank test). When analyzing survival, we found that all infants with AT/RT diagnosed at age <12 months (n = 11 (23%)) died within 18 months from diagnosis, while cases diagnosed at ages 12–35 months (n = 15 (32%)) had a 5-year overall survival probability (5y-OS) of 28%, and those diagnosed at the age of ≥36 months (n = 21 (45%)) had a 5y-OS of 41% (p < 0.0001). All cases treated with surgery only (n = 6 (13%)) died within 6 months; all cases treated with chemotherapy without radiotherapy (n = 12 (25%)) died within 3 years; all cases treated with radiotherapy only (n = 5 (11%)) had a 5y-OS of 60%; the other cases treated with radiotherapy and chemotherapy (n = 24 (51%)) had a 5y-OS of 42.22% (p < 0.0001). Patients with a supratentorial tumor (n = 11 (23%)) had a 5y-OS of 51.95%, those with an unspecified nervous system tumor (n = 14 (30%)) had a 5y-OS of 21.43%, and those with an infratentorial or spine tumor (n = 22 (47%)) had a 5y-OS of 17.36% (p < 0.05).
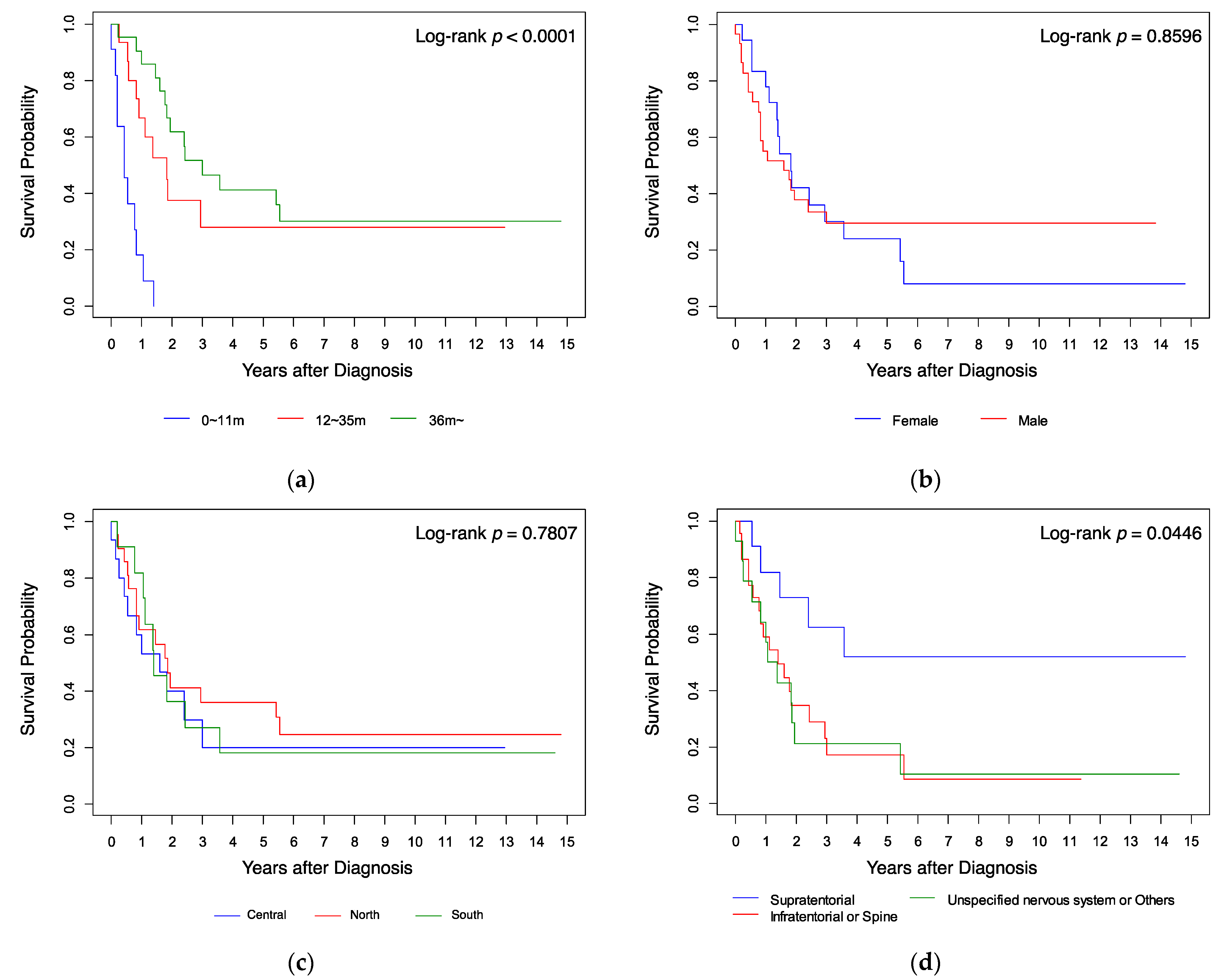 Figure 1. Kaplan–Meier analysis of survival curves compared by (a) age, (b) gender, (c) resident location, (d) tumor site, (e) treatment, and (f) diagnosis year.
Figure 1. Kaplan–Meier analysis of survival curves compared by (a) age, (b) gender, (c) resident location, (d) tumor site, (e) treatment, and (f) diagnosis year.Table 4 shows the results of univariate and multivariate analyses of factors that affect survival. Compared to the age group 0–11 months as a reference, the age groups 12–23 months (HR 0.113, 96% CI 0.039–0.330) and ≥36 months (HR 0.078, 95% CI 0.028–0.216) had a better prognosis on univariate analysis (both p < 0.001). On multivariate analysis, the 12–23 months group had a better prognosis (HR 0.130, 95% CI 0.036–0.468; p = 0.002). Compared to supratentorial tumors, tumors in the infratentorial or spine regions and tumors in an unspecified location of the nervous system or other location had a poorer prognosis in both univariate (HR 3.121, 95% CI 1.146–8.497; p = 0.026 and HR 3.261, 95% CI 1.139–9.337; p = 0.028 respectively) and multivariate analyses (HR 3.234, 95% CI 1.049–9.973; p = 0.041 and HR 3.505, 95% CI 1.121–10.955; p = 0.031). On univariate analysis, chemotherapy (including SCT) (HR 0.079, 95% CI 0.021–0.305; p < 0.001), radiotherapy (HR 0.011, 95% CI 0.002–0.063; p < 0.001), and combined radiotherapy and chemotherapy (HR 0.016, 95% CI 0.004–0.066; p < 0.001) were associated with better outcome. On multivariate analysis, chemotherapy (HR 0.013, 95% CI 0.002–0.097), radiotherapy (HR 0.002, 95% CI 0.000–0.025), and combined radiotherapy and chemotherapy (HR 0.003, 95% CI 0.000–0.031) remained significant protective prognostic factors (all p < 0.001). Gender and diagnosis year were not significant prognostic factors on either univariate or multivariate analysis.
Table 4. Univariate and multivariate analyses of factors affecting outcome.
| Prognostic Factor | Univariate | Multivariate | ||||||
|---|---|---|---|---|---|---|---|---|
| HR | 95% CI | p Value * | HR | 95% CI | p Value * | |||
| Age (months) | ||||||||
| 0–11 m | Reference | Reference | ||||||
| 12–23 m | 0.113 | (0.039–0.330) | <0.001 | 0.130 | (0.036–0.468) | 0.002 | ||
| 24–35 m | 0.379 | (0.082–1.754) | 0.214 | 1.769 | (0.175–17.852) | 0.629 | ||
| ≥36 m | 0.078 | (0.028–0.216) | <0.001 | 0.356 | (0.066–1.929) | 0.231 | ||
| Gender | ||||||||
| Female | Reference | Reference | ||||||
| Male | 0.942 | (0.481–1.844) | 0.862 | 1.012 | (0.430–2.379) | 0.979 | ||
| Tumor site | ||||||||
| Supratentorial | Reference | Reference | ||||||
| Infratentorial or Spine | 3.121 | (1.146–8.497) | 0.026 | 3.234 | (1.049–9.973) | 0.041 | ||
| Unspecified nervous system or Others | 3.261 | (1.139–9.337) | 0.028 | 3.505 | (1.121–10.955) | 0.031 | ||
| Treatment | ||||||||
| Surgery or No treatment | Reference | Reference | ||||||
| Chemotherapy (including SCT) | 0.079 | (0.021–0.305) | <0.001 | 0.013 | (0.002–0.097) | <0.001 | ||
| Radiotherapy | 0.011 | (0.002–0.063) | <0.001 | 0.002 | (0.000–0.025) | <0.001 | ||
| Radiotherapy + Chemotherapy | 0.016 | (0.004–0.066) | <0.001 | 0.003 | (0.000–0.031) | <0.001 | ||
| Diagnosis year | ||||||||
| 1999–2007 | Reference | Reference | ||||||
| 2008–2014 | 1.439 | (0.725–2.856) | 0.298 | 1.316 | (0.477–3.632) | 0.596 | ||
* p values < 0.05 are presented in bold.
3. Conclusions
Since AT/RT is a rare disease, it is not easy for a single center to follow many patients longitudinally. The Taiwan Cancer Registry, a nationwide, population-based database, is therefore a useful resource for monitoring and analyzing the clinical characteristics and the treatment outcomes of AT/RT. It's found that patients at an older age at diagnosis and those with supratentorial tumors had a better prognosis. These data also support the effectiveness of radiotherapy, chemotherapy, or combined radiotherapy and chemotherapy. These data can inform future radiotherapy and chemotherapy regimens, clinical trial design, and risk stratification for AT/RT.
References
- Park, M.; Han, J.W.; Hahn, S.M.; Lee, J.A.; Kim, J.-Y.; Shin, S.H.; Kim, D.-S.; Yoon, H.I.; Hong, K.T.; Choi, J.Y.; et al. Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System in Children Under the Age of 3 Years. Cancer Res. Treat. 2021, 53, 378–388.
- Nesvick, C.L.; Lafay-Cousin, L.; Raghunathan, A.; Bouffet, E.; Huang, A.A.; Daniels, D.J. Atypical Teratoid Rhabdoid Tumor: Molecular Insights and Translation to Novel Therapeutics. J. Neurooncol. 2020, 150, 47–56.
- Ma, X.-J.; Li, D.; Wang, L.; Hao, S.-Y.; Zhang, L.-W.; Zhang, J.-T.; Wu, Z. Overall Survival of Primary Intracranial Atypical Teratoid Rhabdoid Tumor Following Multimodal Treatment: A Pooled Analysis of Individual Patient Data. Neurosurg. Rev. 2020, 43, 281–292.
- Yang, W.-C.; Yen, H.-J.; Liang, M.-L.; Chen, H.-H.; Lee, Y.-Y.; Chang, F.-C.; Lin, S.-C.; Wong, T.-T.; Hu, Y.-W.; Chen, Y.-W. Effect of Early Radiotherapy Initiation and High-Dose Chemotherapy on the Prognosis of Pediatric Atypical Teratoid Rhabdoid Tumors in Different Age Groups. J. Neuro-Oncol. 2020, 147, 619–631.
- Nesvick, C.L.; Nageswara Rao, A.A.; Raghunathan, A.; Biegel, J.A.; Daniels, D.J. Case-Based Review: Atypical Teratoid/Rhabdoid Tumor. Neuro-Oncol. Pract. 2019, 6, 163–178.
- Ostrom, Q.T.; Chen, Y.; de Blank, P.M.; Ondracek, A.; Farah, P.; Gittleman, H.; Wolinsky, Y.; Kruchko, C.; Cohen, M.L.; Brat, D.J.; et al. The Descriptive Epidemiology of Atypical Teratoid/Rhabdoid Tumors in the United States, 2001–2010. Neuro Oncol. 2014, 16, 1392–1399.
- Lau, C.S.; Mahendraraj, K.; Chamberlain, R.S. Atypical Teratoid Rhabdoid Tumors: A Population-Based Clinical Outcomes Study Involving 174 Patients from the Surveillance, Epidemiology, and End Results Database (1973–2010). Cancer Manag. Res. 2015, 7, 301–309.
- Upadhyaya, S.A.; Robinson, G.W.; Onar-Thomas, A.; Orr, B.A.; Johann, P.; Wu, G.; Billups, C.A.; Tatevossian, R.G.; Dhanda, S.K.; Srinivasan, A.; et al. Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-Institutional Trials. Clin. Cancer Res. 2021, 27, 2879.
- Kim, H.Y.; Choi, S.A.; Koh, E.J.; Kim, K.H.; Phi, J.H.; Lee, J.Y.; Kim, S.-K. Combination Treatment of Ci-994 with Etoposide Potentiates Anticancer Effects Through a Topoisomerase ii-Dependent Mechanism in Atypical Teratoid/Rhabdoid Tumor (AT/RT). Front. Oncol. 2021, 11, 648023.
- Oka, H.; Scheithauer, B.W. Clinicopathological Characteristics of Atypical Teratoid/Rhabdoid Tumor. Neurol. Med. Chir. 1999, 39, 510–518.
- Ren, Y.-M.; Wu, X.; You, C.; Zhang, Y.-K.; Li, Q.; Ju, Y. Multimodal Treatments Combined with Gamma Knife Surgery for Primary Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System: A Single-Institute Experience of 18 Patients. Childs Nerv. Syst. 2018, 34, 627–638.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncology 2021, 23, 1231–1251.
- Rao, S.J.B.; Konar, S.K.; Shukla, D.; Bhat, D.I.; Beniwal, M.; Rao, K.V.L.N.; Nandeesh, B.N.; Devi, B.I. Factors Influencing Survival of Children with Atypical Teratoid/Rhabdoid Tumors: A Single-Institute Experience in a Developing Country. World Neurosurg. 2019, 129, e264–e272.
- Li, D.; Heiferman, D.M.; Syed, H.R.; Santos, J.G.; Bowman, R.M.; DiPatri, A.J.; Tomita, T.; Wadhwani, N.R.; Alden, T.D. Pediatric Primary Spinal Atypical Teratoid Rhabdoid Tumor: A Case Series and Review of the Literature. J. Neurosurg. Pediatr. 2019, 24, 267–283.
- Reddy, A.T.; Strother, D.R.; Judkins, A.R.; Burger, P.C.; Pollack, I.F.; Krailo, M.D.; Buxton, A.B.; Williams-Hughes, C.; Fouladi, M.; Mahajan, A.; et al. Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report from the Children’s Oncology Group Trial ACNS0333. J. Clin. Oncol. 2020, 38, 1175–1185.
More
Information
Subjects:
Oncology; Pediatrics
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
883
Revisions:
5 times
(View History)
Update Date:
08 Feb 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No