 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Biljana Mojsoska | + 3389 word(s) | 3389 | 2020-09-03 09:01:08 | | | |
| 2 | Felix Wu | + 3364 word(s) | 3364 | 2020-09-03 10:31:44 | | | | |
| 3 | Felix Wu | Meta information modification | 3364 | 2020-11-03 11:04:16 | | |
Video Upload Options
To design more efficient treatments against bacterial infections, detailed knowledge about the bacterial response to the commonly used antibiotics is required. Proteomics is a well-suited and powerful tool to study molecular response to antimicrobial compounds. Bacterial response profiling from system-level investigations could increase our understanding of bacterial adaptation, the mechanisms behind antibiotic resistance and tolerance development.
1. Introduction
Antimicrobial resistance (AMR) is naturally acquired in microorganisms including bacteria, viruses, fungi, and parasites through mutation or uptake of genetic material. Today AMR is one of the main global health threats. The most urgent problem is AMR in bacteria, and over the years it has been observed that bacteria have developed resistance to every single antibiotic that has come to the market [1]. Due to the misuse of antibiotics, the development and transfer of resistance mechanisms has given rise to multi-drug resistant (MDR) bacteria. In parallel with the antibiotic resistance development, microbial biofilms provide a great platform for an even higher frequency of mutation, thus introducing a greater opportunity for resistance to accumulate and spread [2]. As victims of their success, despite the number of studies that demonstrate the effectiveness of antibiotics, much knowledge on the impact of antibiotics on the overall biological network in bacteria on a system-level is lacking
2. The Role of Proteomic Analysis in Generating New Insight about The Mechanism of Action of Antibiotics and Antibiotic Resistance
With the emergence of antibiotic resistance genes among pathogenic bacteria, proteomic analyses are pivotal in the assessment of the dynamic changes of whole protein expression on a system level. When studying the proteome of pathogenic bacteria there is a high interest to obtain a quantitative view of the differentially expressed proteins in different treatment conditions. Using targeted proteomics one can monitor resistance development and behavior and understand the role of cellular processes when pathogenic bacteria are challenged with antibiotics. In this section, several classes of antibiotics, their mode of action, and studies that report on using proteomic approaches to further explore the bacterial response to these antibiotics will be reviewed. A summary of mechanism of antibiotic resistance is presented in Figure 1.
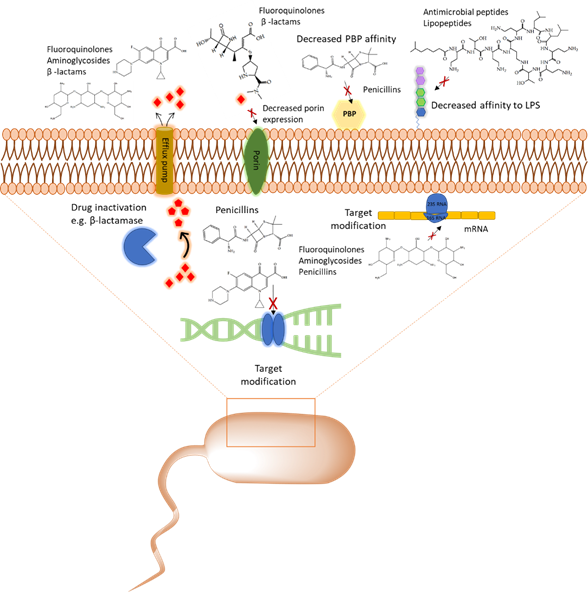
Figure 1. Overview of the common antibiotic resistance mechanism in bacteria. Molecular mechanisms of antibiotic resistance that includes target modification, drug inactivation, decreased affinity to lipopolysaccharides (LPS) and penicillin binding protein (PBP), and expression of porins and efflux pumps are shown.
2.1. Cell Wall Synthesis Inhibitors
Cell wall synthesis inhibitors are a class of antibiotics with the widest use, with Penicillin class (e.g., penicillin G, ampicillin, and oxacillin) being the best known and oldest precursor. Cephalosporins, Cephamycins, Monobactams, and Carbapenems (e.g., meropenem) comprise some of the derivative sub-classes all sharing a β-lactam ring (Figure 2) as a common structural feature [3]. The primary mode of action of this class of antibiotics is inhibition of bacterial cell wall synthesis through binding to an active serine site of penicillin-binding proteins and rendering the enzymes inactive [4] (Figure 1). Several different penicillin-binding proteins can be simultaneously targeted in a single organism affecting different cellular functions [5]. In this way, the enzymes are prevented from catalyzing both the synthesis and cross-linking of peptidoglycan which is essential to achieve a rigid structure. The major lethal consequence is a loss of cell wall integrity and cell death due to osmotic imbalance and/or digestion of the existing cell wall by peptidoglycan hydrolases [6]. However, except for the enzymes that are bound by the antibiotic, more complexes involved in peptidoglycan synthesis are also indirectly affected. This is because of the gradual shortage of peptidoglycan precursors, which makes the cell unable to keep up with the required peptidoglycan synthesis pace to preserve a rigid and efficiently protective wall structure [7].
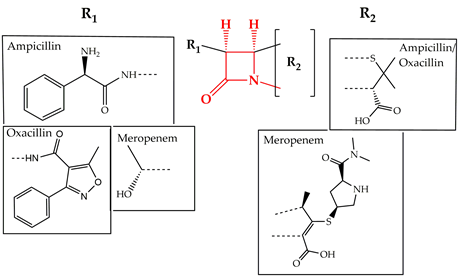
Figure 2. Chemical structure of conserved ring structure in β-lactam antibiotics and side-chain functionalities (R1 and R2) of three β-lactam antibiotics, ampicillin, oxacillin, and meropenem. Dashed lines represent the connecting bonds to the β-lactam ring (red).
Studies on the bacterial response to ampicillin have been done in E. coli. Exposure to sub-MIC of ampicillin together with tetracycline has been used to characterize and compare outer membrane proteome changes in E. coli K12 strain [8]. With eight protein being differentially regulated over ampicillin exposure, ampicillin induced the upregulation of porins OmpC, OmpW, and Tsx, the efflux pump subunit TolC, the usher protein FimD, the Omp assembly factor BamD and the hypothetical lipoprotein YfiO. In this study decreased expression was observed only for the outer membrane protein BamC [8]. In addition, specific protein compounds were associated with outer membrane vesicles-related β-lactam resistance and were also able to confer resistance to susceptible strains in a dose-dependent manner. These proteins included OmpC, OmpF, OmpW, Tolc, and Bcl1. Proteins linked to resistance to different antimicrobial agents (both antibiotics and peptides) were also identified. The suggested resistance mechanism was that the antibiotic could be transferred at a high rate through the overexpressed porins to the lumen of the outer membrane vesicles and get hydrolyzed by β -lactamase enzyme (Figure 1) before being able to act on the cell wall [9]. Mass spectrometry analysis and the Sequential window acquisition of all theoretical mass spectra (SWATH) methods were used to compare the adaptive changes between three different, ciprofloxacin-resistant, carbapenemase-producing Enterobacteriaceae (CPE) strains under meropenem and ciprofloxacin stress. Each antibiotic had caused different proteome alterations and the difference in protein expression has been reported to be more intense in the meropenem-treated cells. Overexpression of the histone-like protein HU, the GroEL/GroES chaperone complex, and the nucleotide exchanging factor Grpe was observed in all carbapenemase-producing strains with the most striking difference in those producing New Delhi metallo-β -lactamase, indicating that DNA and protein stability could be the main factors to be enhanced to increase bacterial fitness. On the contrary, ciprofloxacin only seemed to affect the levels of the outer membrane protein OmpA [10]. Multidrug-resistant sequence type 131 strains have also been targeted through proteomics, in extended-spectrum β lactamase (ESBL)-producing E. coli strains. Five proteins in total were identified and connected to this resistance phenotype, including YahO, YjbJ, YnfD, HdeA, and soluble cytochrome b562, all having an amino acid replaced which was specific for the ST131 sequence type. Through bioinformatics, it was also possible to provide a prediction on how these proteins might interact to contribute to spreading and maintaining the infection in the human intestinal tract. It was suggested that some of the proteins enhance tolerance of the bacteria to gastric acid and some of them prolong the infection through biofilm formation in the intestinal tract [11]. Proteins involved in protein biosynthesis and regulation have also been quantified as the most abundant in clinical strains of E. coli (C538 and C580), producing β-lactamases CMY-2 and TEM-52 [12]. In addition, the MRSA and Methicillin-susceptible S. aureus (MSSA) response to oxacillin, another β-lactam antibiotic, has been investigated using label-free quantitative proteomic strategy. In this study, commonly and differentially expressed proteins had been identified offering complete overview of the antibiotic response in resistant and susceptible bacterial strains [13].
2.2. Inhibitors of Protein Translation
2.2.1. Aminoglycosides
Aminoglycosides, along with tetracyclines and macrolides, are inhibitors of protein translation and represent (Figure 3) a class of antibiotics with a broad activity spectrum that are being used to target infections from both Gram-negative and Gram-positive bacteria, but most commonly those caused by Enterobacteria, such as E. coli [14]. Aminoglycosides exert their activity by inserting themselves into the bacterial cell. This process consists of three consecutive phases [15]. Initially, because of their polycationic nature, aminoglycosides bind through electrostatic interactions to the negatively charged portions of the bacterial membranes [16]. The main bacterial components involved in this interaction are phospholipids and lipopolysaccharides, and teichoic acids in Gram-negative and Gram-positive bacteria, respectively [17]. Displacement of divalent cations is facilitating this interaction with the outer membrane leading to its destabilization. The increased permeability of the membrane facilitates a self-promoted uptake of the antibiotic agents into the periplasmic space [18]. During the 2nd phase, insertion into the cytoplasm is achieved through an energy-dependent manner, that takes advantage of the electron transport chain and membrane potential difference, enabling the insertion of only a few molecules [19]. Interaction of aminoglycosides with specific nucleotides of the 16S bacterial ribosomal RNA creates a barrier to protein synthesis [20]. The outcome is either halting translation or triggering production of mistranslated proteins that can further enhance the drug uptake into the cell by creating membrane pores. The last stage involves higher accumulation levels of the antibiotic in the cell, and hence, a faster pace of translation inhibition as well as mistranslation, explaining the fast and concentration-dependent bacterial killing observed in this particular antibiotic class [21]. Tobramycin, isolated from S. tenebrarius, is a widely used aminoglycoside antibiotic in the clinics and since its first introduction in 1976 [14]. Although the overall class has no strong activity against P. aeruginosa, tobramycin was the most potent agent against it, following amikacin [22]. It has been extensively used alone or in combination with other antibiotics, for treating lung infections in cystic fibrosis patients [23]. Tobramycin, as most aminoglycosides, is a fast-acting bactericidal agent and its action is highly affected by its concentration [22]. Although it has been poorly studied, there is evidence that tobramycin acts through multiple mechanisms. Pharmacodynamic model proposing a dual mechanism of tobramycin against P. aeruginosa with two killing modes: a delayed mode accounting for the effect on protein synthesis (requiring lower concentration) and a more rapid mode corresponding to membrane disruption (requiring higher dosage) has been suggested [24]. Finally, the interaction of tobramycin with the outer membrane can facilitate the entry of additional antibiotics when used in combination with, e.g., β-lactams [25].
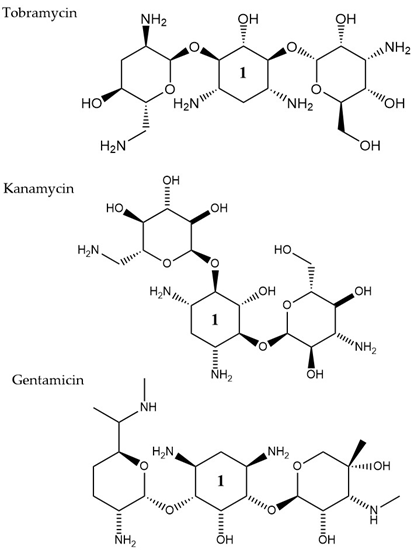
Figure 3. The chemical structures of three aminoglycoside compound. r tobramycin, kanamycin, and gentamicin are shown. All three compounds chare similar ring number 1 indicated across the structures.
LC-MS analysis has further been employed to address the hypothesis that tobramycin could facilitate the eradication of P. aeruginosa infections in cystic fibrosis, through decreasing levels of virulence determinants in secreted outer membrane vesicles. Total proteome analysis of these vesicles revealed 757 proteins, with 66 core proteins also detected in four earlier proteomic studies [26][27][28][29] and 120 proteins were conserved in four P. aeruginosa strains, including two CF clinical ones. Many of the conserved proteins are associated with resistance and virulence, such as efflux pump elements MexA and MexB. After exposure to tobramycin, 165 proteins were downregulated and 17 were up-regulated in the isolated outer membrane vesicles. One of the proteins that were significantly decreased was the virulence factor AprA. This factor is a protease, assumed to indirectly promote dehydration of the airways by inhibiting the secretion of Phe508del-CFTR-Cl- by the epithelial cells of the host and as a result, the clearance of the infection becomes more challenging. The addition of tobramycin- exposed and mutant ΔaprA outer membrane vesicles to bronchial epithelial cells from cystic fibrosis patients led to a decrease in the inhibitory effect of P. aeruginosa on Phe508del-CFTR-Cl- secretion. Finally, significantly lower expression levels were also reported for the virulence factors AlpA/D/E, after tobramycin treatment. All the findings together suggest that tobramycin could improve lung function in cystic fibrosis patients [30]. Adaptive resistance of P. aeruginosa to tobramycin under planktonic conditions has also been investigated through proteomics analysis [31]. The study focused on the evaluation of any proteome changes both, after treatment with a range of sub-MIC concentrations and over a time-course exposure using a fixed tobramycin concentration. Different protein alterations appeared to be clustering together in either higher or lower concentrations. Higher antibiotic dosages induced upregulation of heat shock proteins and proteases, while overexpression of proteins involved in amino acid metabolic/catabolic pathways was observed in treatment using lower drug concentrations. The lowest tobramycin concentration and the highest exposure time induced the wider range of proteome changes among all tested conditions. The most highly up-regulated protein was a heat shock protein (IbpA) that was found to assist cells to resist the effects of the antibiotic by acting together with other proteases, heat shock, and chaperone proteins [31]. In other studies, several proteins of outer membrane vesicles were identified to have a system-level role in antibiotic resistance to other types of aminoglycosides, such as streptomycin [32] and gentamicin [33]. Resistance to Kanamycin, which is another aminoglycoside (Figure 3), through outer membrane proteome modification profiling, was also investigated through mass spectrometry and Western blot. Here, an increase in the levels of TolC, Tsx, and OstA and a decrease in OmpA, FadL OmpW, and MipA, was observed leading the authors to suggest that MipA was a new protein linked to aminoglycoside resistance [34]. Targeted proteomic approaches were employed to study protein pattern alterations that accompany interaction between the two-component system elements CpxA and ArcA in E. coli MG1655 cells when challenged with gentamicin. Here, differentially expressed proteins in wild type and cpxAR mutant strain have been identified including proteins related to metabolism (FruA, FruK, and IlbB) and in stress response (IbpB, IbpA, RpoH, YgiQ, YceA, YncD, YrbL, and DeaD) [35].
2.2.2. Macrolides
There have been three generations of macrolides developed since 1952. The first generation consists of natural components, while the later generations are semi-synthetic and have a wide activity spectrum [36]. The main structural characteristic of this class is a 12–16-member lactone ring with one or more sugars attached (Figure 4). For years, the main mode of action was suggested to be inhibition of protein synthesis merely through association with the 50S ribosomal subunit and obstructing the nascent peptide exit tunnel [37]. Although the precise mode of action remains unclear, several studies over the years have provided insight into new aspects of macrolides’ impact on protein translation. It is now known that macrolides, instead of inducing complete protein synthesis suppression, interfere with specific protein motifs and therefore selectively block peptide elongation [38]. Macrolides can have both bacteriostatic and bactericidal activity [39]. It has been also suggested that the major amino acid sequence responsible for inhibiting translation is Lys/Arg-X-Lys/Arg where both the positive charge and side chains play a role in this process [40].
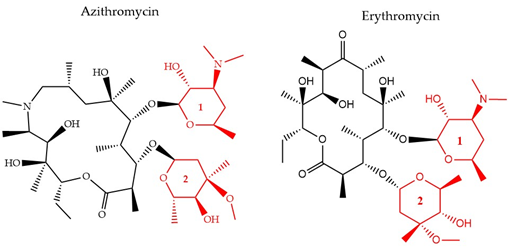
Figure 4. The chemical structure of two antibiotics, azithromycin, and erythromycin that belong to the macrolide class are shown. Conserved ring structures of sugars 1 and 2 (red) and varied lactone ring are shown.
Furthermore, it is hypothesized that besides the recognized sequence mentioned above, there are additional protein fragments, depending on each protein’s sequence, that could modify the peptide’s route in the nascent peptide exit tunnel NAPT and result in decreased efficiency of the antibiotic’s function or differential response to different macrolides [41]. One of the most common resistance mechanisms against macrolides, characterized in S. aureus [42], S. pneumoniae [43], and E. coli [44], is mediated through methylation of the 23S ribosomal domain by methyltransferases encoded by the erythromycin (Figure 3) ribosome methylase gene (erm). The modification leads to a decrease of the affinity between the binding target and the antibiotic, inhibiting their interaction [45] (Figure 1). Active efflux is another way to cope with macrolide-pressure mediated by mefA/E (M-type resistance) and msrD genes in S. pneumoniae [46] and S. pyogenes [47]. Macrolide active efflux has also been characterized in Enterobacteriaceae [48], as well as macrolide-inactivation by phosphorylases and esterases, encoded by ereA/B and mphA/B genes, respectively [44]. The development of resistance in E. coli populations after exposure to sublethal concentrations of erythromycin has been characterized by MALDI-TOF MS and protein radiolabeling. Different exposure times (43, 68, and 103 h) to the antibiotic appeared to lead to the development of different proteome profiles. The proteins that were down-regulated after the shortest exposure time frame were in contrary, up-regulated following the longest treatment. This finding was accompanied by a shift in the subcellular compartment of the up-regulated proteins from the cytoplasm to the outer/inner bacterial membrane. The result of the longest exposure was elevated expression levels in proteins linked to lipid, amino acid, carbohydrate, and polyamine metabolism together with the porin OmpC [33].
2.3. Inhibitors of DNA Synthesis
With the discovery of nalidixic acid in 1962, quinolone development has substantially increased and several antibiotics that share similar structural properties (Figure 5) have been developed since [49]. Fluoroquinolones are especially actively used in the treatment of bacterial infections due to their excellent activity against a wide range of Gram-negative and Gram-positive bacteria. Ciprofloxacin belongs to the broader class of quinolones and is primarily used to address infections caused by Gram-negative bacteria [50]. Among the group of fluoroquinolones, ciprofloxacin has been the most effective agent against P. aeruginosa [51]. This antibiotic acts by binding to DNA gyrase and IV topoisomerase in Gram-negative and Gram-positive bacteria, respectively, and inhibiting DNA replication [52]. This is achieved by stabilization of the DNA-gyrase complex during replication via interaction with the 3-oxo-4-carboxylic acid core (Figure 2) of the antibiotic [53]. The interaction with the DNA is additionally facilitated through the formation of Mg2+ bridges [54]. The association of the complex with ciprofloxacin results in the broken DNA strands being trapped inside the complex and unable to religate. As a consequence, RNA polymerase is additionally blocked, DNA synthesis is halted, and the bacteria die releasing cleaved DNA fragments [55]. A secondary mechanism that contributes to the bactericidal activity of ciprofloxacin is the induction of reactive oxygen species [56]. Ciprofloxacin resistance development has been studied in P. aeruginosa. Although genetic mutations were partly responsible for the adaptive resistance progression, proteomic analysis revealed increased phosphorylation levels of two enzymes, succinate-semialdehyde dehydrogenase (SSADH), and methylmalonate-semialdehyde dehydrogenase (MMSADH). It was hypothesized that these proteins could play a role in ATP production as a supportive mechanism to the efflux pumps. In addition, they could trigger higher nicotinamide adenine dinucleotide phosphate dehydrogenase (NADPH) production to combat the accumulation of hydroxyl radicals induced by ciprofloxacin [57].
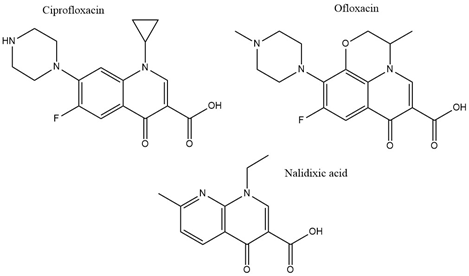
Figure 5. Chemical structures of three clinically used quinolone antibiotics is shown.
Adaptation to ciprofloxacin exposure has also been studied in biofilm cells. Here, the outer membrane proteome modifications in P. aeruginosa biofilms have been identified, after treatment with benzalkonium chloride and ciprofloxacin over 12 consecutive days [58]. Overall, only moderate modifications were reported and the expression levels of the chaperon protein GroEL, the putative tail sheath protein, and the major capsid protein were downregulated in both conditions. Since GroEL would be expected to enhance the flexibility of the bacterium to adapt to the antibiotic stress the opposite results would be anticipated. Regarding the other two proteins, although their precise function is not known it was hypothesized that they might indicate membrane degradation. Finally, ciprofloxacin induced higher levels of the probable bacteriophage protein, which is in agreement with the higher activation rates of bacteriophage genes in biofilms and could be linked to the diverse phenotypes found in a biofilm [58]. Another study looked at different mechanisms involved in a transition from low to high ciprofloxacin resistance through drug pre-exposure in Pseudomonas. The main physiological pathways in each type of resistance were determined using total proteomic analysis of four resistant mutants. Different regulatory pathways were involved in each of the lower resistance levels. Anaerobic respiration was adopted by the mutant with the lowest resistance and the arginine, arginine dehydrogenase, and urease pathways were up-regulated. Slightly higher resistance involved upregulation of proteins having a role in nutrient uptake, such as iron polyamine and amino acids, as well as protein translation. Regarding the mutants demonstrating the highest resistance, the MexCD-oprJ efflux pump had the highest expression levels while proteins involved in quorum sensing were the most downregulated [59]. The later serves as good supporting evidence on many studies that associate efflux pumps as a resistance mechanism to ciprofloxacin [60]. Mass spectrometry experiments confirmed that the intracellular drug concentration was unchanged between wild type cells and low resistant mutants, indicating that the differential regulation of the above-mentioned pathways could be mainly responsible for the resistance [59]. Proteomics has also been applied to selectively analyze subpopulations of P. aeruginosa biofilms that are tolerant to ciprofloxacin. The results showed that the immediate proteome response of antibiotic tolerant subpopulation of P. aeruginosa is characterized by flagellar motility, whereas the adaptive proteome response included upregulation of synthesis of purine molecules related to DNA damage response [61]. Ofloxacin, similar in structure to ciprofloxacin (Figure 2) is a second-line drug used for the treatment of multidrug-resistant tuberculosis (MDR-TB). It has the same target as ciprofloxacin which is DNA gyrase and therefore gyrA and gyrB are often targets for drug resistance. The proteomic analysis shows that there is an increased expression of 14 differentially expressed proteins in TB isolates that carry natural ofloxacin resistance and induced resistance when compared to the isolates susceptible to the drug. Follow up studies on the role of the identified proteins further revealed that few hypothetical proteins might be interfering with the ofloxacin activity by neutralizing the effect of DNA replication, transcription, repair, and recombination [62].
References
- Francesca Prestinaci; Patrizio Pezzotti; Annalisa Pantosti; Antimicrobial resistance: a global multifaceted phenomenon. Pathogens and Global Health 2015, 109, 309-318, 10.1179/2047773215y.0000000030.
- Tim C. R. Conibear; Samuel L. Collins; Jeremy S. Webb; Role of Mutation in Pseudomonas aeruginosa Biofilm Development. PLOS ONE 2009, 4, e6289, 10.1371/journal.pone.0006289.
- Karen Bush; Patricia A. Bradford; β-Lactams and β-Lactamase Inhibitors: An Overview. Cold Spring Harbor Perspectives in Medicine 2016, 6, a025247, 10.1101/cshperspect.a025247.
- Wivagg, C.N.; Bhattacharyya, R.P.; Hung, D.T. Mechanisms of β-lactam killing and resistance in the context of Mycobacterium tuberculosis. J. Antibiot. (Tokyo) 2014, 67, 645–654.
- Tomasz, A. The Mechanism of the Irreversible Antimicrobial Effects of Penicillins: How the Beta-Lactam Antibiotics Kill and Lyse Bacteria. Annu. Rev. Microbiol. 1979, 33, 113–137.
- Tipper, D.J. Mode of action of β-lactam antibiotics. Rev. Infect. Dis. 1979, 1, 39–53.
- Cho, H.; Uehara, T.; Bernhardt, T.G. Beta-lactam antibiotics induce a lethal malfustioning of the bacterial cell wall sunthesis machinery. Cell 2014, 159, 1300–1311.
- Changxin Xu; Xiangmin Lin; Haixia Ren; Yueling Zhang; Sanying Wang; Xuanxian Peng; Analysis of outer membrane proteome ofEscherichia coli related to resistance to ampicillin and tetracycline. PROTEOMICS 2006, 6, 462-473, 10.1002/pmic.200500219.
- Si Won Kim; Seong Bin Park; Se Pyeong Im; Jung Seok Lee; Jae Wook Jung; Tae Won Gong; Jassy Mary S. Lazarte; Jaesung Kim; Jong-Su Seo; Jong-Hwan Kim; et al.Jong-Wook SongHyun Suk JungGwang Joong KimYoung Ju LeeSuk-Kyung LimTae Sung Jung Outer membrane vesicles from β-lactam-resistant Escherichia coli enable the survival of β-lactam-susceptible E. coli in the presence of β-lactam antibiotics. Scientific Reports 2018, 8, 1-13, 10.1038/s41598-018-23656-0.
- Hanna E. Sidjabat; Jolene Gien; David Kvaskoff; Keith Ashman; Kanchan Vaswani; Sarah Reed; Ross P. McGeary; David L. Paterson; Amanda Bordin; Gerhard Schenk; et al. The use of SWATH to analyse the dynamic changes of bacterial proteome of carbapanemase-producing Escherichia coli under antibiotic pressure. Scientific Reports 2018, 8, 1-11, 10.1038/s41598-018-21984-9.
- Akihiro Nakamura; Masaru Komatsu; Yuki Ohno; Nobuyoshi Noguchi; Akira Kondo; Naoya Hatano; Identification of specific protein amino acid substitutions of extended-spectrum β-lactamase (ESBL)-producing Escherichia coli ST131: a proteomics approach using mass spectrometry. Scientific Reports 2019, 9, 1-8, 10.1038/s41598-019-45051-z.
- Gilberto Igrejas; Luís Pinto; Patrícia Poeta; Hajer Radhouani; Céline Coelho; Carlos Augusto Brandao De Carvalho; Jorge Rodrigues; Carmen Torres; Rui Vitorino; Pedro Domingues; et al. Proteomic evaluation of Escherichia coli isolates from human clinical strains. Journal of Integrated OMICS 2011, 1, 42-48, 10.5584/jiomics.v1i1.20.
- Xiaofen Liu; Yingwei Hu; Pei-Jing Pai; Daijie Chen; Henry Hei Ning Lam; Label-Free Quantitative Proteomics Analysis of Antibiotic Response inStaphylococcus aureusto Oxacillin. Journal of Proteome Research 2014, 13, 1223-1233, 10.1021/pr400669d.
- Kevin M. Krause; Alisa W. Serio; Timothy R. Kane; Lynn E. Connolly; Aminoglycosides: An Overview. Cold Spring Harbor Perspectives in Medicine 2016, 6, a027029, 10.1101/cshperspect.a027029.
- † Andrew Thompson; ‡ Jürgen Schäfer; ‡ Karsten Kuhn; ‡ Stefan Kienle; ‡ Josef Schwarz; † Günter Schmidt; ‡ And Thomas Neumann; ‡ Christian Hamon; Tandem Mass Tags: A Novel Quantification Strategy for Comparative Analysis of Complex Protein Mixtures by MS/MS. Analytical Chemistry 2003, 75, 1895-1904, 10.1021/ac0262560.
- Hancock, R.E.W.; Farmer, S.W.; Li, Z.; Poolet, K. Interaction of Aminoglycosides with the Outer Membranes and Purified Lipopolysaccharide and OmpF Porin of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1309–1314.
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.M.Y.S. Bacterial Uptake of Aminoglycoside Antibiotics. J. Antimicrob. Chemother. 1987, 51, 439–457.
- Jana, S.; Deb, J.K. Molecular understanding of aminoglycoside action and resistance. Appl. Microbiol. Biotechnol. 2006, 70, 140–150.
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Updat. 2010, 13, 151–171.
- Tsai, A.; Uemura, S.; Johansson, M.; Puglisi, E.V.; Marshall, R.A.; Aitken, C.E.; Korlach, J.; Ehrenberg, M.; Puglisi, J.D. The Impact of Aminoglycosides on the Dynamics of Translation Elongation. Cell Rep. 2013, 3, 497–508.
- Davis, B.D.; Chen, L.; Tai, P.C. Misread protein creates membrane channels: An essential step in the bactericidal action of aminoglycosides. Proc. Natl. Acad. Sci. USA 1986, 83, 6164–6168.
- Serio, A.W.; Keepers, T.; Andrews, L.; Krause, K.M. Aminoglycoside Revival: Review of a Historically Important Class of Antimicrobials Undergoing Rejuvenation. EcoSal Plus 2018, 8.
- Shteinberg, M.; Elborn, J.S. Use of Inhaled Tobramycin in Cystic Fibrosis. Adv. Ther. 2015, 32, 1–9.
- Bulitta, J.B.; Ly, N.S.; Landersdorfer, C.B.; Wanigaratne, N.A.; Velkov, T.; Yadav, R.; Oliver, A.; Martin, L.; Shin, B.S.; Forrest, A.; et al. Two mechanisms of killing of pseudomonas aeruginosa by tobramycin assessed at multiple inocula via mechanism-based modeling. Antimicrob. Agents Chemother. 2015, 59, 2315–2327.
- Yadav, R.; Bulitta, J.B.; Schneider, E.K.; Shin, B.S.; Velkov, T.; Nation, R.L.; Landersdorfer, C.B. Aminoglycoside Concentrations Required for Synergy with Carbapenems against Pseudomonas aeruginosa Determined via Mechanistic Studies and Modeling. Antimicrob. Agents Chemother. 2017, 61, 1–16.
- Park, A.J.; Murphy, K.; Surette, M.D.; Bandoro, C.; Krieger, J.R.; Taylor, P.; Khursigara, C.M. Tracking the Dynamic Relationship between Cellular Systems and Extracellular Subproteomes in Pseudomonas aeruginosa Biofilms. J. Proteome Res. 2015, 14, 4524–4537.
- Reales-Calderón, J.A.; Corona, F.; Monteoliva, L.; Gil, C.; Martínez, J.L. Quantitative proteomics unravels that the post-transcriptional regulator Crc modulates the generation of vesicles and secreted virulence determinants of Pseudomonas aeruginosa. J. Proteomics 2015, 127, 352–364.
- Toyofuku, M.; Roschitzki, B.; Riedel, K.; Eberl, L. Identification of Proteins Associated with the Pseudomonas aeruginosa Biofilm Extracellular Matrix. J. Proteome Res. 2012, 11, 4906–4915.
- Choi, D.-S.; Kim, D.-K.; Choi, S.J.; Lee, J.; Choi, J.-P.; Rho, S.; Park, S.-H.; Kim, Y.-K.; Hwang, D.; Gho, Y.S. Proteomic analysis of outer membrane vesicles derived from Pseudomonas aeruginosa. Proteomics 2011, 11, 3424–3429.
- Katja Koeppen; Roxanna Barnaby; Angelyca A. Jackson; Scott A. Gerber; Deborah A. Hogan; Bruce A. Stanton; Tobramycin reduces key virulence determinants in the proteome of Pseudomonas aeruginosa outer membrane vesicles. PLoS ONE 2019, 14, e0211290, 10.1371/journal.pone.0211290.
- Xia Wu; Kiara Held; Chunxiang Zheng; Benjamin J. Staudinger; Juan D. Chavez; Chad R. Weisbrod; Jimmy K. Eng; Pradeep K. Singh; Colin Manoil; James E. Bruce; et al. Dynamic Proteome Response of Pseudomonas aeruginosa to Tobramycin Antibiotic Treatment. Molecular & Cellular Proteomics 2015, 14, 2126-2137, 10.1074/mcp.m115.050161.
- Hui Li; Bao-Cheng Wang; Wen-Jiao Xu; Xiang-Min Lin; Xuan-Xian Peng; Identification and Network of Outer Membrane Proteins Regulating Streptomysin Resistance inEscherichia coli. Journal of Proteome Research 2008, 7, 4040-4049, 10.1021/pr800310y.
- Denisa Petráčková; Jiří Janeček; Silvia Bezoušková; Ladislava Kalachová; Zuzana Techniková; Karolína Buriánková; Petr Halada; Kateřina Haladová; Jaroslav Weiser; Fitness and proteome changes accompanying the development of erythromycin resistance in a population of Escherichia coli grown in continuous culture. MicrobiologyOpen 2013, 2, 841-852, 10.1002/mbo3.121.
- Dan-Feng Zhang; Hui Li; Xiang-Min Lin; Xuan-Xian Peng; Outer membrane proteomics of kanamycin-resistant Escherichia coli identified MipA as a novel antibiotic resistance-related protein. FEMS Microbiology Letters 2015, 362, 1-8, 10.1093/femsle/fnv074.
- Emina Ćudić; Kristin Surmann; Gianna Panasia; Elke Hammer; Sabine Hunke; The role of the two-component systems Cpx and Arc in protein alterations upon gentamicin treatment in Escherichia coli. BMC Microbiology 2017, 17, 1-17, 10.1186/s12866-017-1100-9.
- George P Dinos; The macrolide antibiotic renaissance. Journal of Cerebral Blood Flow & Metabolism 2017, 174, 2967-2983, 10.1111/bph.13936.
- Anna Janas; Piotr Przybylski; 14- and 15-membered lactone macrolides and their analogues and hybrids: structure, molecular mechanism of action and biological activity. European Journal of Medicinal Chemistry 2019, 182, 111662, 10.1016/j.ejmech.2019.111662.
- Vázquez-Laslop, N.; Mankin, A.S. How Macrolide Antibiotics Work. Trends Biochem. Sci. 2018, 43, 668–684.
- Svetlov, M.S.; Vázquez-Laslop, N.; Mankin, A.S. Kinetics of drug-ribosome interactions defines the cidality of macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2017, 114, 13673–13678.
- Sothiselvam, S.; Neuner, S.; Rigger, L.; Klepacki, D.; Micura, R.; Vázquez-Laslop, N.; Mankin, A.S. Binding of Macrolide Antibiotics Leads to Ribosomal Selection against Specific Substrates Based on Their Charge and Size. Cell Rep. 2016, 16, 1789–1799.
- Davis, A.R.; Gohara, D.W.; Yap, M.-N.F. Sequence selectivity of macrolide-induced translational attenuation. Proc. Natl. Acad. Sci. USA 2014, 111, 15379–15384.
- Yao, W.; Xu, G.; Li, D.; Bai, B.; Wang, H.; Cheng, H.; Zheng, J.; Sun, X.; Lin, Z.; Deng, Q.; et al. Staphylococcus aureus with an erm-mediated constitutive macrolide-lincosamide-streptogramin B resistance phenotype has reduced susceptibility to the new ketolide, solithromycin. BMC Infect. Dis. 2019, 19, 175.
- Schroeder, M.R.; Stephens, D.S. Macrolide Resistance in Streptococcus pneumoniae. Front. Cell. Infect. Microbiol. 2016, 6, 98.
- Gomes, C.; Ruiz-Roldán, L.; Mateu, J.; Ochoa, T.J.; Ruiz, J. Azithromycin resistance levels and mechanisms in Escherichia coli. Sci. Rep. 2019, 9, 6089.
- Fyfe, C.; Grossman, T.H.; Kerstein, K.; Sutcliffe, J. Resistance to Macrolide Antibiotics in Public Health Pathogens. Cold Spring Harb. Perspect. Med. 2016, 6, a025395.
- Chancey, S.T.; Zhou, X.; Zähner, D.; Stephens, D.S. Induction of efflux-mediated macrolide resistance in Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2011, 55, 3413–3422.
- Oggioni, M.R.; Rossolini, G.M.; Stefani, S.; et al. Type M Resistance to Macrolides Is Due to a Two-Gene Efflux Transport System of the ATP-Binding Cassette (ABC) Superfamily. Front. Microbiol. 2018, 9.
- Chollet, R.; Chevalier, J.; Bryskier, A.; Pagès, J.-M. The AcrAB-TolC pump is involved in macrolide resistance but not in telithromycin efflux in Enterobacter aerogenes and Escherichia coli. Antimicrob. Agents Chemother. 2004, 48, 3621–3624.
- Monique I. Andersson; Development of the quinolones. Journal of Antimicrobial Chemotherapy 2003, 51, 1-11, 10.1093/jac/dkg212.
- Marc Lebel; Ciprofloxacin: Chemistry, Mechanism of Action, Resistance, Antimicrobial Spectrum, Pharmacokinetics, Clinical Trials, and Adverse Reactions. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 1988, 8, 3-30, 10.1002/j.1875-9114.1988.tb04058.x.
- Rehman, A.; Patrick, W.M.; Lamont, I.L. Mechanisms of ciprofloxacin resistance in pseudomonas aeruginosa: New approaches to an old problem. J. Med. Microbiol. 2019, 68, 1–10.
- Naqvi, S.A.R.; Roohi, S.; Iqbal, A.; Sherazi, T.A.; Zahoor, A.F.; Imran, M. Ciprofloxacin: From infection therapy to molecular imaging. Mol. Biol. Rep. 2018, 45, 1457–1468.
- Zhang, G.F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612.
- Palumbo, M.; Gatto, B.; Zagotto, G.; Palu, G. On the mechanism of action of quinolone drugs. Bull. Johns Hopkins Hosp. 1993, 1, 232–235.
- Drlica, K.; Zhao, X. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 1997, 61, 377–392.
- Jensen, P.Ø.; Briales, A.; Brochmann, R.P.; Wang, H.; Kragh, K.N.; Kolpen, M.; Hempel, C.; Bjarnsholt, T.; Høiby, N.; Ciofu, O. Formation of hydroxyl radicals contributes to the bactericidal activity of ciprofloxacin against Pseudomonas aeruginosa biofilms. Pathog. Dis. 2014, 70, 440–443.
- Hsun-Cheng Su; Kevin Ramkissoon; Janet Doolittle; Martha Clark; Jainab Khatun; Ashley Secrest; Matthew C. Wolfgang; Morgan C. Giddings; The Development of Ciprofloxacin Resistance in Pseudomonas aeruginosa Involves Multiple Response Stages and Multiple Proteins. Antimicrobial Agents and Chemotherapy 2010, 54, 4626-4635, 10.1128/aac.00762-10.
- Idalina Machado; Laurent Coquet; Proteomic Changes in Pseudomonas aeruginosa Biofilm Cells after Adaptive Resistance Development. Journal of Proteomics & Bioinformatics 2016, 9, null, 10.4172/jpb.1000390.
- Jianhe Peng; Jing Cao; Fui Mee Ng; Jeffrey Hill; Pseudomonas aeruginosa develops Ciprofloxacin resistance from low to high level with distinctive proteome changes. Journal of Proteomics 2017, 152, 75-87, 10.1016/j.jprot.2016.10.005.
- Jinsong Zhou; Dongyun Hao; Xudong Wang; Teimei Liu; Chengyan He; Feng Xie; Yanhong Sun; Jin Zhang; An important role of a “probable ATP-binding component of ABC transporter” during the process ofPseudomonas aeruginosa resistance to fluoroquinolone. PROTEOMICS 2006, 6, 2495-2503, 10.1002/pmic.200501354.
- Brett M. Babin; Lydia Atangcho; Mark B. Van Eldijk; Michael J. Sweredoski; Annie Moradian; Sonja Hess; Tim Tolker-Nielsen; Dianne K. Newman; David A. Tirrell; Selective Proteomic Analysis of Antibiotic-Tolerant Cellular Subpopulations in Pseudomonas aeruginosa Biofilms. mBio 2017, 8, e01593-17, 10.1128/mbio.01593-17.
- Manju Lata; Divakar Sharma; Nirmala Deo; Pramod Kumar Tiwari; Deepa Bisht; Krishnamurthy Venkatesan; Proteomic analysis of ofloxacin-mono resistant Mycobacterium tuberculosis isolates. Journal of Proteomics 2015, 127, 114-121, 10.1016/j.jprot.2015.07.031.

