Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Christian Quintus Scheckhuber | + 3489 word(s) | 3489 | 2022-01-25 04:25:34 | | | |
| 2 | Christian Quintus Scheckhuber | Meta information modification | 3489 | 2022-01-25 14:43:45 | | | | |
| 3 | Rita Xu | Meta information modification | 3489 | 2022-01-26 02:22:06 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Scheckhuber, C. Superoxide Dismutases. Encyclopedia. Available online: https://encyclopedia.pub/entry/18774 (accessed on 27 July 2026).
Scheckhuber C. Superoxide Dismutases. Encyclopedia. Available at: https://encyclopedia.pub/entry/18774. Accessed July 27, 2026.
Scheckhuber, Christian. "Superoxide Dismutases" Encyclopedia, https://encyclopedia.pub/entry/18774 (accessed July 27, 2026).
Scheckhuber, C. (2022, January 25). Superoxide Dismutases. In Encyclopedia. https://encyclopedia.pub/entry/18774
Scheckhuber, Christian. "Superoxide Dismutases." Encyclopedia. Web. 25 January, 2022.
Copy Citation
Several enzymes exist that can convert or degrade reactive oxygen species (ROS); among them are the superoxide dismutases (SODs). SODs are responsible for converting superoxide anions to hydrogen peroxide by dismutation, therefore participating in the ROS detoxification. Here a short overview on the role of SODs in development and pathogenicity of fungi like Podospora anserina and Aspergillus spp. is given.
Aspergillus
development
pathogenicity
Podospora anserina
reactive oxygen species
1. Introduction
Superoxide dismutases (SOD) are antioxidant metalloenzymes that dismutate O2− into molecular oxygen and hydrogen peroxide (H2O2), thus, eliminating superoxide radicals. They are key players in defending cells from reactive oxygen species (ROS) during an infection of pathogens [1]. ROS, like superoxide, hydrogen peroxide, hydroxyl radicals, and singlet oxygen, among others, are usually toxic to the cell in elevated concentrations because of their high reactivity with biologically relevant molecules, e.g., proteins, nucleic acids, and lipids. However, in physiological concentrations, some of them are also important regulators of cellular signaling processes. Oxidative stress and its careful management are also important as it is one of the most common means of defense employed by the immune system when combating invading pathogens. As such, ROS are components of the direct and indirect antimicrobial immune response [2]. Furthermore, SODs can be regarded as a crucial defense against pathogens that survive attacks by the immune system of the host during an infection. Therefore, they tend to be the ideal drug target for different therapies [3]. In evolutionary terms, SODs show little structural homology, indicating the convergent evolution of this group of enzymes [4][5]. SODs even have different functions depending on their location within the cell and the organism.
The filamentous ascomycete P. anserina serves as a model for aging while the other organisms are mostly pathogenic. Various species of Aspergilli are grouped and discussed (Figure 1). At this point we would like to note that the SOD nomenclature is not standardized across the literature, and our work reflects this fact. In naming SODs, we generally use the nomenclature found in the references.
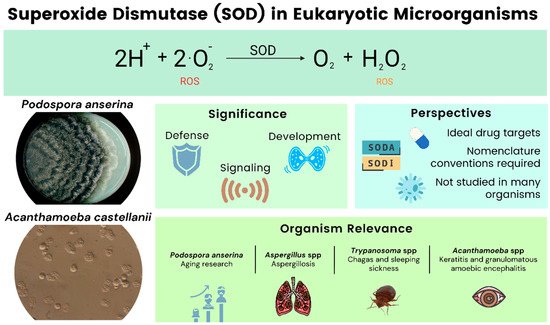
Figure 1. Overview of SODs in eukaryotic microorganisms.
2. Fungi
In this section, the findings on oxidative stress in general, and SODs in particular, from a variety of fungi are reported. Although not a pathogen, research on P. anserina shows an impact on human health and disease by its use as a valuable model for studying the process of biological aging. A variety of Aspergillus species are being reviewed, as oxidative stress and antioxidant defense are the key regulators for the pathogenicity of most of these molds.
2.1. Podospora anserina: A Model System for Biological Aging
Biological aging is defined as the continuous loss of vitality and viability, with a concomitant increase in morbidity and mortality [6]. Although there have been tremendous efforts to understand the aging process, there is still no consensus on how aging and its unwanted ‘side-effects’ can be reduced or countered. Rather, simple model systems have been utilized to foster our understanding of how processes, such as senescence and aging, mechanistically work. Fungi, for example, are attractive organisms on which to study the time-dependent changes that lead, ultimately, to organismal death [7][8]. In addition to the unicellular baker’s yeast (Saccharomyces cerevisiae), a considerable effort has been invested into characterizing aging at the molecular and cellular levels in the filamentous ascomycete, P. anserina, which is a close relative of the more well-known Neurospora crassa [9][10][11][12]. Among the studied pathways, the formation and detoxification of harmful ROS in P. anserina are particularly well-characterized [13][14]. According to the Free Radical Theory of Aging, ROS are causal agents of the aging process [15][16]. Although P. anserina is an obligate aerobe, mitochondrial defects are not necessarily lethal. In addition to the conventional complex IV (Cytochrome c oxidase, COX), the fungus can use an alternative oxidase (AOX) for maintaining its viability in case COX activity becomes compromised [17]. AOX-dependent respiration was found to lead to the decreased production of ROS in plants and fungi [18][19]. Supporting this line of evidence is the observation that numerous long-lived mutants of P. anserina utilize AOX, but not COX, as terminal oxidase [17][20]. In this section, we will focus on the role of SODs on P. anserina development (Figure 2).
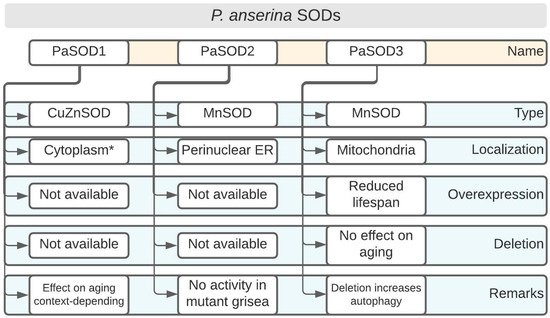
Figure 2. Podospora anserina SOD overview. * Further cellular localizations such as intermembrane space of mitochondria are likely. Overexpression and deletion denote genetic manipulations of the corresponding SOD-encoding genes. ER: endoplasmic reticulum.
2.1.1. PaSOD1: A Differentially Regulated CuZnSOD in Certain Long-Lived Mutants
Two long-lived P. anserina mutants, that display interesting differences regarding their SOD activity profiles, will be briefly discussed here. The findings show that, depending on the genetic context, these enzymes might be potentially linked to a lifespan extension. SOD activity was assessed in the mutants, grisea and ex1, and was compared to the wild type (WT), which exhibits a normal lifespan [17][21]. In the WT, the activity of the mostly cytoplasmic CuZnSOD, PaSOD1, (Figure 2) increased strongly with age, whereas the activity of PaSOD2, a MnSOD which is either ER-associated or secreted [22], decreased [17]. The mutant ex1, in which AOX was induced due to a deletion in the mtDNA that led to the loss of the CoI gene (encoding the first subunit of COX), seemed to exclusively employ PaSOD1 [21]. In mutant grisea, a gene encoding a copper-modulated transcription factor (GRISEA) is functionally inactivated by mutation [23]. This mutant experienced severely depleted cellular copper levels [24]. Therefore, the activity of copper-containing proteins, such as COX and PaSOD1, is almost undetectable [21][25]. Importantly, putative target genes of the transcription factor GRISEA are, at most, very weakly expressed. Among these is PaCtr3 (encoding a high-affinity copper transporter of the plasma membrane), which contributes to the severely reduced copper levels in grisea. In the WT, the differential activity of PaSOD1 pointed to a redistribution of cellular copper during aging where the amount of available copper in the cytoplasm increased. This is an example of a clear developmental regulation of PaSOD1 activity at the protein level.
In addition, PaSOD1 seems to play a crucial role in extension of the lifespan of the PaCox17::ble mutant [26]. In this mutant, the putative copper chaperone, PaCOX17, was deleted by gene replacement. The mutant displays an increased lifespan compared to the WT; even after 320 days, 40 out of the 60 cultures were still alive, whereas WT isolates have a lifespan of two to three weeks. PaSOD1 levels are highly upregulated in PaCox17::ble, suggesting an improved defense against superoxide anions [26].
2.1.2. PaSOD2: The Enigmatic MnSOD Is Inactivated in the Long-Lived Mutant grisea
Not all SODs are characterized equally in P. anserina. Here, PaSOD2 is introduced, which offers interesting perspectives for future research to elucidate its potential roles in the developmental processes of P. anserina, such as aging.
PaSOD2 has an apparent molecular weight of 26.8 kDa and it contains putative manganese/iron binding motifs [22]. Originally, PaSOD2 was reported to represent a mitochondrial SOD [21][25], although it lacks a clear mitochondrial targeting sequence [22]. Later work showed that PaSOD2 is present in the perinuclear endoplasmic reticulum [22] (Figure 2). The role of a mitochondrial SOD is taken by PaSOD3 [22], which will be discussed below.
As mentioned previously, PaSOD2 activity decreases during aging in P. anserina WT strains [17]. PaSod2 was originally thought to be a putative target gene of the copper-modulated transcription factor, GRISEA, because it is not detected in isolates of the mutant [17]. Due to the redistribution of cellular copper during the aging of P. anserina, GRISEA was supposed to be inactivated, leading to transcriptional inactivation of PaSod2 [21]. Later research using transcriptional profiling showed that PaSod2 was transcribed at WT levels in mutant grisea when copper was added to the growth medium; thus, it is not a target gene of GRISEA [27].
2.1.3. PaSOD3: Increasing the Content of This Mitochondrial MnSOD, but Not Its Ablation, Has Effects on Aging
This SOD is, by far, the best studied of the three identified family members of P. anserina (Figure 2). Mutants in which PaSod3 levels were experimentally modulated revealed some unexpected findings, while cleary illustrating that PaSOD3 is linked to other important surveillance and quality control systems in P. anserina. PaSOD3 might also influence several processes by modulating ROS levels (superoxide anion and hydrogen peroxide) that are required for signaling, underscoring the role of ROS as cellular messengers.
PaSOD3 constitutes a mitochondrial SOD with a deduced molecular weight of 25.5 kDa. The enzyme contains a putative mitochondrial targeting sequence and was experimentally demonstrated to reside in mitochondria [22]. The impact of PaSOD3 on aging was studied by utilizing mutants in which PaSod3 was either constitutively overexpressed or deleted [22]. ∆PaSod3 strains were hypersensitive to compounds that generated superoxide anions, such as paraquat, which was expected. In contrast, the sensitivity to hydrogen peroxide was not altered in ∆PaSod3. However, the overexpression of PaSod3 led to a significant growth reduction compared to the WT control, even without any additional stressors. Unexpectedly, these strains were more sensitive to both paraquat and hydrogen peroxide when these compounds were added to the growth medium [22]. Regarding the aging process, ∆PaSod3 did not show any significant differences compared to the WT control. On the other hand, PaSod3 overexpressors were short-lived when grown under standard conditions, reaching only around 75% of the median lifespan of the WT [22]. This can be explained, firstly, by the protein levels of the peroxiredoxin PaPRX1, a mitochondrial peroxidase that detoxifies hydrogen peroxide which was strongly reduced in PaSod3 overexpressors, suggesting a compromised defense against ROS. Secondly, several proteins responsible for the quality control of mitochondria, such as matrix proteases PaCLPP and PaLON, were almost absent, or they exhibited altered protein patterns, due to their incomplete processing or degradation, respectively [22]. Thirdly, the mitochondrial heat shock protein PaHSP60 was clearly upregulated and proteolytically activated in the PaSod3 overexpressing strains [22]. The authors suggest that the elevated PaSOD3 levels led to the generation of high doses of hydrogen peroxide in mitochondria, which was subsequently converted to the highly reactive hydroxyl radical. The latter is formed by Fenton chemistry involving hydrogen peroxide and various metal ions and it has a highly destructive potential due to its extremely high reactivity with lipids, nucleic acids, and proteins [28].
To better understand the phenotypic effects of the increased PaSod3 expression, a modeling strategy was employed that supported the hypothesis that excess hydrogen peroxide generated by PaSOD3 was responsible for the damaging effects [29]. The computational studies suggested that the levels of the PaSOD3 cofactor, Mn2+, were elevated by a factor of 80 in the PaSod3 overexpressors. This result led to a study of the role of manganese supplementation of the growth medium of the WT and the PaSod3 overexpressors on phenotypic parameters [30]. Interestingly, the supplementation of the growth medium with MnSO4, even in small amounts (20 µM), led to a reversion of the PaSod3_OEx mutant phenotype. In addition to showing WT-like growth rates, the PaSod3 overexpressors had restored the formation of aerial hyphae and fertility. Regarding aging, MnSO4 concentrations of 80 µM allowed the PaSod3_OEx strains to reach median lifespans indistinguishable from those of the WTs [30].
Importantly, the quantification of the total SOD activity (PaSOD1, PaSOD2 and PaSOD3) demonstrated no significant differences in whole cell extracts or isolated mitochondria, regardless of whether Mn was added to the growth medium or not. The authors concluded that a general limitation of manganese on SOD activity in PaSod3 overexpressors did not occur [30].
However, in contrast to the WT, the Mn-supplemented PaSod3 overexpressors demonstrated elevated levels of peroxidase and catalase activities in addition to the upregulated protein levels of the peroxiredoxin, PaPRX. Further results suggested that a hitherto unknown Mn-dependent protein contributed to an improved degradation rate of hydrogen peroxide [30].
PaSOD3 protein levels were shown to be controlled by the protein, PaRCF1 [31]. PaRCF1 belongs to the HIG1 (‘hypoxia-inducible gene 1’) family of proteins, which play an important role in the assembly and organization of the mitochondrial respiratory chain [32]. A deleted mutant of PaRcf1, ∆PaRcf1, contains only a fraction of PaSOD3 proteins (approximately 20%) compared to the WT, but its activity appears not to be affected [31]. However, ∆PaRcf1 is hypersensitive to the addition of the redox cycler, paraquat, into the medium in comparison to the WT. Additionally, this mutant fails to maintain a normal growth rate, is sterile, and is marked by a decreased median lifespan. It is important to note that in ∆PaRcf1, PaSOD3, as well as other factors of maintenance and cellular quality control decreased, e.g., PaLON, PaPRX and PaCLP [31]. Thus, the altered phenotype of ∆PaRcf1 is likely the result of several deficiencies.
As mentioned above, the signaling function of ROS is altered in PaSod3 deletion strains [30]. It is known that ROS are modulators of various cellular processes; among these is the controlled cellular ’self-eating’, or autophagy [33][34]. Usually, ROS are activators of autophagy, which acts as a system to assist in maintaining cellular homeostasis [35]. It was shown that the ’unexpected healthy phenotype’ of ∆PaSod3 was due to the induction of autophagy (Figure 2) [36]. The authors demonstrated this by analyzing the number of autophagosomes using the marker Gfp-PaAtg8. These increased, even in juvenile stages. Increased autophagy (especially mitophagy) is important for ∆PaSod3 survival because the ∆PaSod3 ∆Paatg1 double knockout strain reveals severe phenotypic deficiencies, such as reductions in both the growth rate and lifespan [36].
SODs are enzymes that degrade superoxide anions. However, the generation of superoxide anions is an important process for growth and development in fungi. P. anserina and many other organisms express genes that encode plasma membrane NADPH oxidases, which are sources of superoxide production [37][38].
It was found that genes encoding NADPH oxidases (Nox) are exclusively present in the genomes of multicellular organisms, regardless of their phylogenetic origin, pointing to a role of controlled superoxide production in differentiation processes [39]. Deactivating the P. anserina gene, PaNox1, leads to several defects, such as the reduced pigmentation of the mycelium, the insufficient formation of aerial hyphae and, most importantly, the compromised formation of perithecia (fruiting bodies). PaNOX1 functions in cellular signaling upstream of the mitogen-activated protein kinase kinase kinase (MAPKKK), PaASK1 [40]. Meanwhile, the deletion of PaNox2, a second NADPH oxidase isoform in P. anserina, demonstrates that this gene is essential for the germination of ascospores [40]. In summary, the regulated secretion of superoxide anions and peroxide during the life cycle is controlled by proteins, including PaNOX1, PaNOX2 and PaASK1 [40].
Enzymes such as PaSOD2 are potentially secreted, degrading superoxide anions, thereby modulating the O2−-mediated developmental signaling, although, for now, this is speculative and requires further investigation.
The work on the role of SODs in the developmental process of P. anserina firmly positions these proteins as crucial components of an elaborate network, controlling several vital processes, such as growth, fertility, the intracellular communication of biological quality control pathways and, ultimately, aging.
2.2. Aspergillus spp.: The Often Pathogenic Fungi Affecting Human Health
The genus Aspergillus contains several hundred members. Most of them have a cosmopolitan distribution. Many species of this genus are known for their pathogenicity, e.g., A. fumigatus and A. flavus. Others display only mild pathogenicity and are of industrial importance, such as A. niger, A. oryzae, and A. terreus. A. nidulans is a valuable model organism for studying eukaryotic cell biology.
2.2.1. A. fumigatus: One of the Most Important Fungal Pathogens
A. fumigatus is one of the best characterized fungal pathogens. In the environment, it plays a crucial role in the recycling of carbon and nitrogen. Due to its abundant sporulation, it releases a huge quantity of conidia into the air that is potentially problematic for people with a compromised immune system. Aspergillosis symptoms first begin in the lungs. In severely predisposed individuals, any organ can be targeted [41]. A. fumigatus is “the most important opportunistic human pathogen among phylogenetically closely related aspergilli” [42]. SOD research using A. fumigatus has revealed several important aspects on how these enzymes co-modulate pathogenicity. Several key findings are outlined below.
The A. fumigatus genome encodes four putative SODs: AfSOD1p, a cytoplasmic CuZnSOD; AfSOD2p, a mitochondrial MnSOD; AfSOD3p, a cytoplasmic MnSOD; and AfSOD4p, which displays a MnSOD domain at its C-terminus [43].
The genes encoding AfSOD1p and AfSOD2p are strongly expressed in A. fumigatus conidia. In contrast, AfSOD3p is found mostly in the mycelium. Although the gene encoding AfSOD4p is only weakly expressed, its deletion is lethal to the fungus. However, it remains to be experimentally shown as to whether AfSOD4p is a bona fide SOD or not [43]. Research showed that the decreased resistance to temperature stress and the ROS-generating menadione are the hallmarks of AfSOD1 and AfSOD2 deletion. The construction of a triple deletion mutant, ∆AfSOD1-3, resulted in a complex phenotype. The deletion strains were very sensitive to menadione and were killed by the macrophages in the alveoli of immunocompetent mice. Unexpectedly, virulence, per se, was not distinguishable from the control parental strain in immunocompromised mice [43]. Copper insertion is a critical step for CuZnSOD maturation. In baker’s yeast, this step is mediated by the copper chaperone, Ccs1p [44]. Recently, the role of the Ccs1p ortholog from A. fumigatus has been studied in detail [45]. The resulting mutant of a deletion of ccsA was characterized by the elevated accumulation of ROS and a higher degree of sensitivity to oxidative stress. Importantly, the conserved CXC motif was essential for the interaction between CcsA, SODA, and the adaptation to oxidative stress. SODC, a MnSOD, complemented the defects exhibited by ccsA or sodA deletions, if overexpressed, or when Mn2+ was added to the growth medium [45]. Interestingly, the abrogation of the CcsA–SODA complex did not lead to an altered virulence of the fungus.
In addition to building blocks of the cell wall, such as galactomannan, chitin synthetases, and immune response modulators (rodA/hyp1 and pksP/alb1), several proteins mediating oxidative stress defense have been implicated in pathogenicity, e.g., catalases (Cat1p and Cat2p) and SODs (MnSOD and CuZnSOD) [46].
In A. fumigatus, the expression of genes encoding SODs were found to be controlled by the regulator of the G-protein signaling protein, RgsC, which is highly conserved in ascomycetes [47]. Furthermore, the deletion of the rgsC gene led to a pleiotropic phenotype. The described hallmarks include compromised growth, asexual development, the reduced ROS tolerance of conidia, and the decreased virulence in the wax moth [47]. The phenotype of the mutant could be related to the compromised ROS signaling because the gene expressions and enzymatic activities of catalases and SODs were severely decreased [47].
Under the conditions of iron-deprivation, A. fumigatus was found to transcriptionally upregulate genes encoding CuZnSOD and other iron-independent antioxidant proteins [42][48]. This is not unexpected, because several important antioxidant enzymes depend on the levels of available iron as a cofactor, such as catalases and heme peroxidases [49]. Conditions of low iron and oxidative stress are encountered by A. fumigatus when it colonizes the human body [42]. It was found, through a transcriptome analysis, that iron deprivation increases oxidative stress susceptibility and pharmacologically targeting these iron-independent antioxidant defense systems might offer the potential to effectively combat fungal infections. Purified CuZnSOD from A. fumigatus has been successfully employed in the detection of aspergillosis because it has a high level of immune reactivity to the sera of patients [50].
2.2.2. A. flavus: Spoiling Food by Toxin Production
A. flavus is a human pathogen that can cause aspergillosis. Additionally, it can spoil several food sources such as cereal grains, legumes, and tree nuts. The consumption of contaminated feed can increase the chance for liver cancer because A. flavus produces the mycotoxin, Aflatoxin B1 [51]. Aflatoxin B1 is one of the most dangerous mycotoxins. Several results point towards a connection between aflatoxin synthesis and the production and detoxification of ROS. Aflatoxin B1 toxicity can be ameliorated by piperine, which is usually found in certain peppers [52][53]. It was shown that piperine not only downregulated the set of genes that led to aflatoxin toxicity, but it also positively influenced the concentrations of antioxidants in A. flavus [54]. Several genes that encode antioxidant proteins, including members of the SOD family, were significantly upregulated upon piperine treatment. There is a clear correlation between levels of ROS and the capability of A. flavus to synthesize aflatoxin [55][56][57]. However, a study showed that the superoxide generator, menadione, was capable of decreasing aflatoxin production in A. flavus NRRL3357 [58]. In this study, SOD activity decreased in the presence of menadione. A study aimed at deciphering the interplay between oxidative stress, superoxide activity, and aflatoxin biosynthesis found that the redox cycler and the superoxide generator, paraquat, at low concentrations, inhibited aflatoxin production [59]. The transcription of the regulator gene aflR and the aflatoxin biosynthetic cluster genes were downregulated by the paraquat treatment. Although the addition of purified CuZnSOD to the culture medium counteracted the paraquat-mediated increase in superoxide production by A. flavus, aflatoxin biosynthesis was not restored, because the CuZnSOD protein itself displayed an inhibitory effect [59]. By contrast, cytosolic and mitochondrial superoxide production was capable of downregulating aflatoxin production by decreasing aflR expression. The mechanism by which supplementing CuZnSOD to the growth medium resulted in a reduction of aflatoxin formation is not yet clear, but it likely does not involve the detoxification of superoxide anions [59].
References
- Maurya, R.; Namdeo, M. Superoxide dismutase: A key enzyme for the survival of intracellular pathogens in host. In Reactive Oxygen Species; Ahmad, R., Ed.; IntechOpen Limited: London, UK, 2021.
- Herb, M.; Schramm, M. Functions of ROS in macrophages and antimicrobial immunity. Antioxidants 2021, 10, 313.
- Paiva, C.N.; Bozza, M.T. Are reactive oxygen species always detrimental to pathogens? Antioxid. Redox Signal. 2013, 20, 1000–1037.
- Case, A.J. On the origin of superoxide dismutase: An evolutionary perspective of superoxide-mediated redox signaling. Antioxidants 2017, 6, 82.
- Landis, G.N.; Tower, J. Superoxide dismutase evolution and life span regulation. Mech. Ageing Dev. 2005, 126, 365–379.
- Jazwinski, S.M.; Kim, S. Examination of the dimensions of biological age. Front. Genet. 2019, 10, 263.
- Kaeberlein, M. Lessons on longevity from budding yeast. Nature 2010, 464, 513–519.
- Bhattacharya, S.; Bouklas, T.; Fries, B.C. Replicative aging in pathogenic fungi. J. Fungi 2021, 7, 6.
- Lorin, S.; Dufour, E.; Sainsard-Chanet, A. Mitochondrial metabolism and aging in the filamentous fungus Podospora anserina. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 604–610.
- Scheckhuber, C.Q.; Osiewacz, H.D. Podospora anserina: A model organism to study mechanisms of healthy ageing. Mol. Genet. Genom. 2008, 280, 365–374.
- van Diepeningen, A.D.; Engelmoer, D.J.P.; Sellem, C.H.; Huberts, D.H.E.W.; Marijke Slakhorst, S.; Sainsard-Chanet, A.; Zwaan, B.J.; Hoekstra, R.F.; Debets, A.J.M. Does autophagy mediate age-dependent effect of dietary restriction responses in the filamentous fungus Podospora anserina? Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130447.
- Osiewacz, H.D.; Schürmanns, L. A network of pathways controlling cellular homeostasis affects the onset of senescence in Podospora anserina. J. Fungi 2021, 7, 263.
- Gredilla, R.; Grief, J.; Osiewacz, H.D. Mitochondrial free radical generation and lifespan control in the fungal aging model Podospora anserina. Exp. Gerontol. 2006, 41, 439–447.
- Wiemer, M.; Osiewacz, H.D. Effect of paraquat-induced oxidative stress on gene expression and aging of the filamentous ascomycete Podospora anserina. Microb. Cell 2014, 1, 225–240.
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300.
- Son, J.M.; Lee, C. Aging: All roads lead to mitochondria. Semin. Cell Dev. Biol. 2021, 116, 160–168.
- Borghouts, C.; Werner, A.; Elthon, T.; Osiewacz, H.D. Copper-modulated gene expression and senescence in the filamentous fungus Podospora anserina. Mol. Cell. Biol. 2001, 21, 390–399.
- Minagawa, N.; Koga, S.; Nakano, M.; Sakajo, S.; Yoshimoto, A. Possible involvement of superoxide anion in the induction of cyanide-resistant respiration in Hansenula anomala. FEBS Lett. 1992, 302, 217–219.
- Maxwell, D.P.; Wang, Y.; McIntosh, L. The alternative oxidase lowers mitochondrial reactive oxygen production in plant cells. Proc. Natl. Acad. Sci. USA 1999, 96, 8271–8276.
- Scheckhuber, C.Q.; Houthoofd, K.; Weil, A.C.; Werner, A.; de Vreese, A.; Vanfleteren, J.R.; Osiewacz, H.D. Alternative oxidase dependent respiration leads to an increased mitochondrial content in two long-lived mutants of the ageing model Podospora anserina. PLoS ONE 2011, 6, e16620.
- Borghouts, C.; Scheckhuber, C.Q.; Werner, A.; Osiewacz, H.D. Respiration, copper availability and SOD activity in P. anserina strains with different lifespan. Biogerontology 2002, 3, 143–153.
- Zintel, S.; Schwitalla, D.; Luce, K.; Hamann, A.; Osiewacz, H.D. Increasing mitochondrial superoxide dismutase abundance leads to impairments in protein quality control and ROS scavenging systems and to lifespan shortening. Exp. Gerontol. 2010, 45, 525–532.
- Osiewacz, H.D.; Nuber, U. GRISEA, a putative copper-activated transcription factor from Podospora anserina involved in differentiation and senescence. Mol. Gen. Genet. 1996, 252, 115–124.
- Borghouts, C.; Osiewacz, H.D. GRISEA, a copper-modulated transcription factor from Podospora anserina involved in senescence and morphogenesis, is an ortholog of MAC1 in Saccharomyces cerevisiae. Mol. Gen. Genet. 1998, 260, 492–502.
- Borghouts, C.; Scheckhuber, C.Q.; Stephan, O.; Osiewacz, H.D. Copper homeostasis and aging in the fungal model system Podospora anserina: Differential expression of PaCtr3 encoding a copper transporter. Int. J. Biochem. Cell Biol. 2002, 34, 1355–1371.
- Stumpferl, S.W.; Stephan, O.; Osiewacz, H.D. Impact of a disruption of a pathway delivering copper to mitochondria on Podospora anserina metabolism and life span. Eukaryot. Cell 2004, 3, 200–211.
- Servos, J.; Hamann, A.; Grimm, C.; Osiewacz, H.D. A differential genome-wide transcriptome analysis: Impact of cellular copper on complex biological processes like aging and development. PLoS ONE 2012, 7, e49292.
- Wardman, P.; Candeias, L.P. Fenton chemistry: An introduction. Radiat. Res. 1996, 145, 523–531.
- Kowald, A.; Hamann, A.; Zintel, S.; Ullrich, S.; Klipp, E.; Osiewacz, H.D. A systems biological analysis links ROS metabolism to mitochondrial protein quality control. Mech. Ageing Dev. 2012, 133, 331–337.
- Grimm, C.; Osiewacz, H.D. Manganese rescues adverse effects on lifespan and development in Podospora anserina challenged by excess hydrogen peroxide. Exp. Gerontol. 2015, 63, 8–17.
- Fischer, F.; Filippis, C.; Osiewacz, H.D. RCF1-dependent respiratory supercomplexes are integral for lifespan-maintenance in a fungal ageing model. Sci. Rep. 2015, 5, 12697.
- Strogolova, V.; Furness, A.; Robb-McGrath, M.; Garlich, J.; Stuart, R.A. Rcf1 and Rcf2, members of the hypoxia-induced gene 1 protein family, are critical components of the mitochondrial cytochrome bc1-cytochrome c oxidase supercomplex. Mol. Cell. Biol. 2012, 32, 1363–1373.
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427.
- Wang, Y.; Nartiss, Y.; Steipe, B.; McQuibban, G.A.; Kim, P.K. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy 2012, 8, 1462–1476.
- Priault, M.; Salin, B.; Schaeffer, J.; Vallette, F.M.; di Rago, J.P.; Martinou, J.C. Impairing the bioenergetic status and the biogenesis of mitochondria triggers mitophagy in yeast. Cell Death Differ. 2005, 12, 1613–1621.
- Knuppertz, L.; Warnsmann, V.; Hamann, A.; Grimm, C.; Osiewacz, H.D. Stress-dependent opposing roles for mitophagy in aging of the ascomycete Podospora anserina. Autophagy 2017, 13, 1037–1052.
- Vignais, P.V. The superoxide-generating NADPH oxidase: Structural aspects and activation mechanism. Cell. Mol. Life Sci. 2002, 59, 1428–1459.
- Bokoch, G.M.; Knaus, U.G. NADPH oxidases: Not just for leukocytes anymore! Trends Biochem. Sci. 2003, 28, 502–508.
- Lalucque, H.; Silar, P. NADPH oxidase: An enzyme for multicellularity? Trends Microbiol. 2003, 11, 9–12.
- Malagnac, F.; Lalucque, H.; Lepère, G.; Silar, P. Two NADPH oxidase isoforms are required for sexual reproduction and ascospore germination in the filamentous fungus Podospora anserina. Fungal Genet. Biol. 2004, 41, 982–997.
- Latgé, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350.
- Kurucz, V.; Krüger, T.; Antal, K.; Dietl, A.M.; Haas, H.; Pócsi, I.; Kniemeyer, O.; Emri, T. Additional oxidative stress reroutes the global response of Aspergillus fumigatus to iron depletion. BMC Genom. 2018, 19, 357.
- Lambou, K.; Lamarre, C.; Beau, R.; Dufour, N.; Latge, J.P. Functional analysis of the superoxide dismutase family in Aspergillus fumigatus. Mol. Microbiol. 2010, 75, 910–923.
- Culotta, V.C. Superoxide dismutase, oxidative stress, and cell metabolism. Curr. Top. Cell. Regul. 2001, 36, 117–132.
- Du, W.; Zhai, P.; Liu, S.; Zhang, Y.; Lua, L. The copper chaperone CcsA, coupled with superoxide dismutase SodA, mediates the oxidative stress response in Aspergillus fumigatus. Appl. Environ. Microbiol. 2021, 87, 1–16.
- Rementeria, A.; López-Molina, N.; Ludwig, A.; Vivanco, A.B.; Bikandi, J.; Pontón, J.; Garaizar, J. Genes and molecules involved in Aspergillus fumigatus virulence. Rev. Iberoam. Micol. 2005, 22, 1–23.
- Kim, Y.; Heo, I.B.; Yu, J.H.; Shin, K.S. Characteristics of a regulator of G-protein signaling (RGS) rgsC in Aspergillus fumigatus. Front. Microbiol. 2017, 8, 2058.
- Oberegger, H.; Zadra, I.; Schoeser, M.; Haas, H. Iron starvation leads to increased expression of Cu/Zn-superoxide dismutase in Aspergillus. FEBS Lett. 2000, 485, 113–116.
- Dlouhy, A.C.; Outten, C.E. The iron metallome in Eukaryotic organisms. Met. Ions Life Sci. 2013, 12, 241–278.
- Holdom, M.D.; Lechenne, B.; Hay, R.J.; Hamilton, A.J.; Monod, M. Production and characterization of recombinant Aspergillus fumigatus Cu,Zn superoxide dismutase and its recognition by immune human sera. J. Clin. Microbiol. 2000, 38, 558–562.
- Amaike, S.; Keller, N.P. Aspergillus flavus. Annu. Rev. Phytopathol. 2011, 49, 107–133.
- Madhyastha, M.S.; Bhat, R.V. Aspergillus parasiticus growth and aflatoxin production on black and white pepper and the inhibitory action of their chemical constituents. Appl. Environ. Microbiol. 1984, 48, 376–379.
- Lee, S.E.; Mahoney, N.E.; Campbell, B.C. Inhibition of aflatoxin B1 biosynthesis by piperlongumine isolated from Piper longum L. J. Microbiol. Biotechnol. 2002, 12, 679–682.
- Caceres, I.; El Khoury, R.; Bailly, S.; Oswald, I.P.; Puel, O.; Bailly, J.D. Piperine inhibits aflatoxin B1 production in Aspergillus flavus by modulating fungal oxidative stress response. Fungal Genet. Biol. 2017, 107, 77–85.
- Narasaiah, K.V.; Sashidhar, R.B.; Subramanyam, C. Biochemical analysis of oxidative stress in the production of aflatoxin and its precursor intermediates. Mycopathologia 2006, 162, 179–189.
- Fountain, J.C.; Scully, B.T.; Chen, Z.Y.; Gold, S.E.; Glenn, A.E.; Abbas, H.K.; Lee, R.D.; Kemerait, R.C.; Guo, B. Effects of hydrogen peroxide on different toxigenic and atoxigenic isolates of Aspergillus flavus. Toxins 2015, 7, 2985–2999.
- Fountain, J.C.; Koh, J.; Yang, L.; Pandey, M.K.; Nayak, S.N.; Bajaj, P.; Zhuang, W.J.; Chen, Z.Y.; Kemerait, R.C.; Lee, R.D.; et al. Proteome analysis of Aspergillus flavus isolate-specific responses to oxidative stress in relationship to aflatoxin production capability. Sci. Rep. 2018, 8, 3430.
- Zaccaria, M.; Ludovici, M.; Sanzani, S.M.; Ippolito, A.; Cigliano, R.A.; Sanseverino, W.; Scarpari, M.; Scala, V.; Fanelli, C.; Reverberi, M. Menadione-induced oxidative stress re-shapes the oxylipin profile of Aspergillus flavus and its lifestyle. Toxins 2015, 7, 4315–4329.
- Furukawa, T.; Sakuda, S. Inhibition of aflatoxin production by paraquat and external superoxide dismutase in Aspergillus flavus. Toxins 2019, 11, 107.
More
Information
Subjects:
Biochemistry & Molecular Biology; Biology; Microbiology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
894
Revisions:
3 times
(View History)
Update Date:
06 Feb 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No