Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Samuele Ciceri | + 1585 word(s) | 1585 | 2021-12-01 04:56:52 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Ciceri, S. Inhibitor-Enzyme Complexes for New Anti-TB Agents. Encyclopedia. Available online: https://encyclopedia.pub/entry/18654 (accessed on 24 June 2026).
Ciceri S. Inhibitor-Enzyme Complexes for New Anti-TB Agents. Encyclopedia. Available at: https://encyclopedia.pub/entry/18654. Accessed June 24, 2026.
Ciceri, Samuele. "Inhibitor-Enzyme Complexes for New Anti-TB Agents" Encyclopedia, https://encyclopedia.pub/entry/18654 (accessed June 24, 2026).
Ciceri, S. (2022, January 23). Inhibitor-Enzyme Complexes for New Anti-TB Agents. In Encyclopedia. https://encyclopedia.pub/entry/18654
Ciceri, Samuele. "Inhibitor-Enzyme Complexes for New Anti-TB Agents." Encyclopedia. Web. 23 January, 2022.
Copy Citation
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB), is the most devastating human pathogen, as confirmed by the latest TB Report published in October.
tuberculosis
structure-based drug design
fragment-based drug design
1. Introduction
The protracted antibiotic therapy, even if cheap and with high cure rates among treated patients, presents a number of negative factors and is hindered by many issues, including the phenomenon of persistence, the drug resistance, and the co-infection with HIV. Once inhaled into the lungs, the bacterium is phagocytosed by macrophages, where it encounters an environment limited in many nutrients, such as sugars and amino acids. Under these nutritionally limited conditions, enzymes essential for the growth and the survival of Mtb represent attractive targets for the design of new potential drugs.
Considering the increasing difficulty of modern-day drugs to effectively tackle TB, many researchers are directing their efforts toward the discovery of new medicines.
Understanding enzyme inhibition processes can help conceptualize different types of inhibitors and characterize potential anti-mycobacterial pathways/mechanisms. Exploiting the structural information allows the thorough evaluation of diverse compounds, providing the knowledge required to efficiently optimize leads toward differentiated candidate drugs.
2. Mtb Enzymatic Targets
A selection of structural studies (2018–present) on chemical entities exhibiting anti-TB properties and acting on specific Mtb enzymes is here reported with the intent to highlight the most promising candidates and their key contacts within the binding site of the target. The understanding of the ligand-enzyme interaction is fundamental to rationalize medicinal chemistry efforts in the discovery and development of new drugs, to predict potential off-target interactions with human counterparts, and to evaluate the likelihood of possible transfers of resistance factors among bacteria.
2.1. Isocitrate Lyase 1 (ICL1)
Fatty acids have been shown to be a major source of carbon and energy for Mtb in clinically infected lungs. Two catabolic pathways have been described for their utilization: the β-oxidation cycle and the glyoxylate shunt. The glyoxylate shunt circumvents the loss of two carbon dioxides from the tricarboxylic acid (TCA) cycle, thereby permitting the net incorporation of carbon into cellular structures during growth on acetate. This mechanism is widespread among prokaryotes, lower eukaryotes, and plants but it is absent in vertebrates. The glyoxylate shunt is particularly important for the persistence of various infectious agents, including Mtb. Interestingly, an upregulation of isocitrate lyases (ICL, isoform 1 and 2) has been observed in bacteria and fungi during infection [1]. ICLs are key enzymes converting isocitrate/methylisocitrate to glyoxylate/pyruvate and succinate in the glyoxylate shunt and methylcitrate cycle, respectively (Scheme 1) [2]. Hence, because ICLs play a crucial role in both persistence and virulence, they can be considered ideal targets for the development of new anti-TB agents [1].
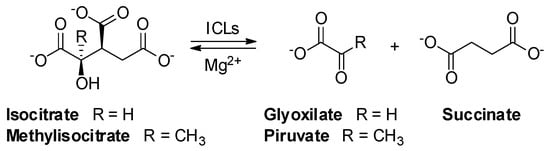
Scheme 1. Reversible lysis reaction catalyzed by ICLs in the glyoxylate shunt: conversion of isocitrate to glyoxylate and succinate.
For this purpose, the 3D structures of Mtb ICL1 in the apo form (PDB: 1F61) and in complex with the inhibitors 3-bromopyruvate (1), 3-nitropropionate (2), and itaconate (3) (Figure 1) were studied by means of X-ray diffraction (XRD). In the host, 3 is synthesized by decarbonylation of cis-aconitate upon activation of immune cells [3]. Despite the enzyme acts as a tetramer, it is also stable in solution as a dimer. The monomeric structure of ICL1 consists of sixteen α-helices, five 310-helices, twelve β-strands, and several loops; the active sites are located near the interface of the two subunits.
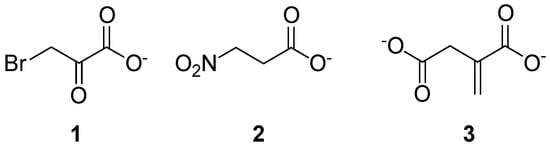
Figure 1. Chemical structure of compounds 1–3.
The inhibition mechanism of 1 is based on a dehalogenation, followed by the formation of a covalent adduct with the active site nucleophile Cys191; supposedly, this modification produces conformational changes in the L5 loop and C-terminal tail, ultimately leading to the closure of the binding region. The most recent structure of ICL1 in complex with 3 (IC50 = 420 μM) showed that the inhibitor was bound to the active site of the enzyme (Figure 2, PDB: 6XPP). In detail, the hydrogen bonding interactions between the carboxylate moieties of the inhibitor and Asn313, Ser315, Ser317, Thr347, Arg228, and Mg2+ were found to be essential for the binding. Adding hydrophobic, aromatic, and polar moieties to the alkene of 3 significantly lowered the inhibition potency, suggesting that its proximity to Cys191 is a prerequisite for the covalent interaction to occur. The crystal structure of the ICL-3 complex revealed that the enzyme was in its closed conformation, consistently with previous data. This finding confirmed that the binding of the compound to the active site is crucial to induce conformational changes in regions that control access to the binding pocket [4].
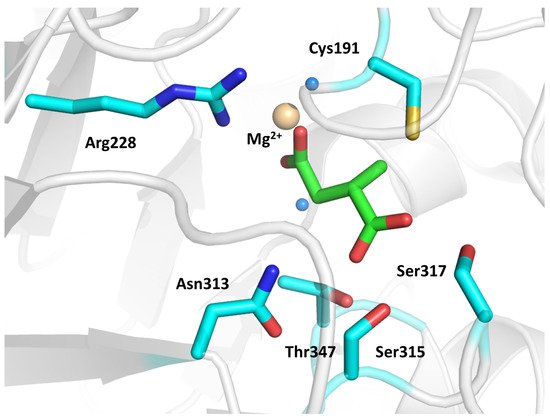
Figure 2. Crystal structure of 3 in complex with ICL1 (PDB: 6XPP). The ligand and interacting residues are represented as green and cyan sticks, respectively. The Mg2+ ion is shown as a golden sphere, while ordered water molecules are visualized as blue spheres.
The crystallographic information provided important insights into the mechanism of ICL1, suggesting that the itaconate scaffold can be utilized to develop new selective inhibitors, branching out at the C3 position of 3 to fill the spacious pocket occupied by glyoxylate.
2.2. Methylmalonyl-CoA Mutase (MCM)
Mycobacterial B12-dependent methylmalonyl-CoA mutase (MCM) catalyzes the reversible isomerization of methylmalonyl-CoA (M-CoA) to succinyl-CoA via a 5′-deoxyadenosylcobalamin (AdoCbl)-dependent radical mechanism (Figure 3).
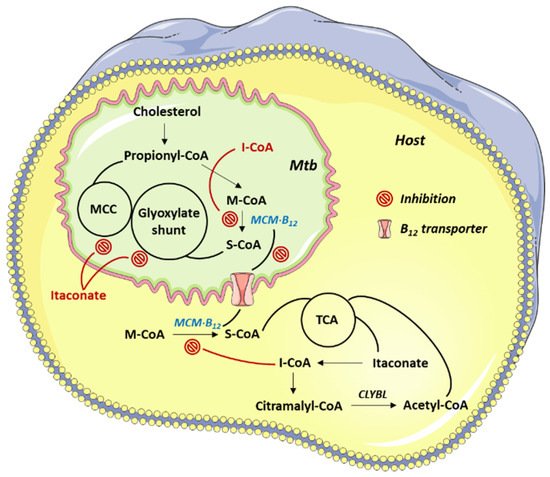
Figure 3. Mtb pathways targeted by 4.
The coenzyme A (CoA) derivative of 3, itaconyl-CoA (I-CoA, 4), is a suicide inactivator of the human and mycobacterial MCMs. Because MCM plays an essential role in lipid breakdown, inhibition studies on this enzyme have recently been reported [5]. The mechanisms underlying 4-mediated MCM inhibition were studied through the EPR and MS analyses of the reaction products between the 5′-deoxyadenosyl radical (dAdo•) and 4. The authors concluded that dAdo• added to the double bond of 4 affording a tertiary carbon radical, stabilized by delocalization onto the adjacent carboxylate (Scheme 2).
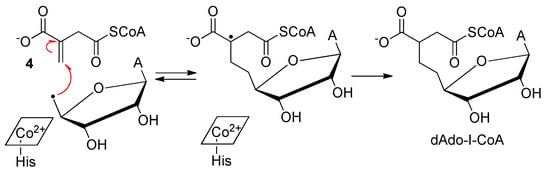
Scheme 2. Reaction between dAdo• and 4.
A crystallographic study of AdoCbl-MCM complexed with 4 allowed to characterize the binding between the enzyme and the inhibitor (Figure 4). In the native heterodimeric structure, AdoCbl inserted its dimethylbenzimidazole tail in a side pocket, while His265 served as the lower axial ligand. On the opposite face, the 5′-carbon of dAdo was 2.5 Å away from the cobalt atom; the adenine was coplanar with the corrin ring and oriented above the pyrrole rings. The binding of 4 induced a large conformational change in the AdoCbl α unit, which collapsed around the binding pocket.
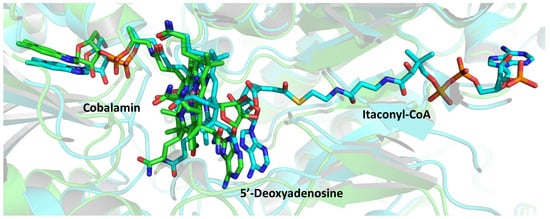
Figure 4. Superimposition of the active sites of native MCM (green, PDB: 6OXC) and of its complex with 4 (cyan, PDB: 6OXD). The ligands are represented as colored sticks.
The authors concluded that the homolysis of the cobalt-carbon bond in AdoCbl generated dAdo• and a spectator cobalamin radical; compound 4 triggered this cleavage, mimicking a normal catalytic cycle, but proximity effects promoted suicidal addition of dAdo• to its double bond.
The demonstration that 4 is an inhibitor of MCM is consistent with the reported observation that the incidence of active TB is markedly lower in patients affected by pernicious anemia, which causes B12 deficiency [6].
2.3. Fumarate Hydratase
Fumarate hydratase (fumarase), a component of the citric acid cycle, is essential for Mtb survival. This enzyme catalyzes the reversible hydration of fumarate to malate (Scheme 3), and its depletion has been linked to an impaired mycobacterial growth due to the accumulation of fumarate, which reacts with cysteine thiols. The resulting oxidative stress causes the death of the microorganism both in vitro and in vivo. Unfortunately, targeting fumarase poses a significant challenge because the protein is highly evolutionarily conserved. In particular, the human and Mtb homologs share identical active site residues, as well as a 53% overall sequence identity.
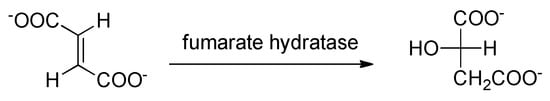
Scheme 3. Reversible lysis reaction catalyzed by fumarase.
The mycobacterial enzyme is a 210 kDa class-II fumarase, featuring a homotetrameric structure. Each subunit is characterized by an N-terminal domain (residues 1–137), a central domain (residues 138–393), and a flexible C-terminal domain (residues 394–474). The protein has four active sites, located in a cleft formed by three subunits, and covered by a loop that allows the switch between “open” and “closed” states.
In 2016, the discovery of an inhibitor, compound 5 (IC50 = 2.5 μM), was reported [7]. The selectivity of the molecule was due to its binding to an allosteric pocket, composed of unconserved residues located on two C-terminal domains of the fumarase tetramer (Figure 5).
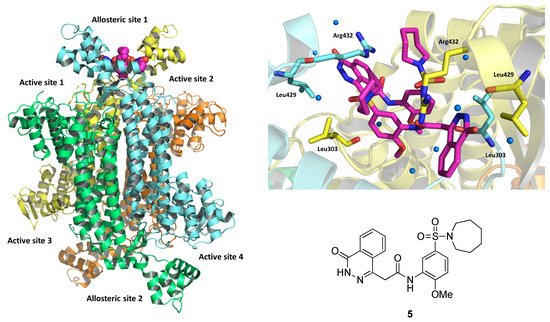
Figure 5. On the left, depiction of the X-ray crystal structure of the fumarase-5 complex (PDB: 5F91). The four chains are evidenced in different colors, and the two bound inhibitors are represented as magenta spheres. On the top right, expansion of the binding site, showing the ligand as magenta sticks. Interacting residues are represented as yellow and light-blue sticks depending on the chain to which they belong. On the bottom right, chemical structure of 5.
Despite 5 did not exhibit bactericidal activity, it was able to inhibit the growth of the mycobacterium; hence, additional investigations were carried out in 2019 by the same research group [8]. In detail, 5 was deconstructed into “fragment-like” molecules, affording a library of compounds with a molecular weight in the range 204–326 Da. However, this defragmentation approach proved to be unsuccessful, as evidenced by both biochemical assays and differential scanning fluorimetry (DSF) tests. Therefore, a traditional structure–activity relationship (SAR) study was undertaken. A series of compounds, resulting from modifications of the different moieties of 5, was prepared and screened against the enzyme. The authors identified two benzoazepanyl derivatives, 6 and 7, showing a three-fold stronger inhibition compared to 5, and a set of compounds (8–12, Figure 6) endowed with a measurable minimal inhibitory concentration (MIC) against Mtb in vitro (6.3–9 μM in 7H9/DPPC). All derivatives maintained the dimeric binding mode to the allosteric site of the enzyme, as confirmed by XRD. These encouraging results confirm the importance of fumarase as a target for the development of bacteriostatic compounds against TB.
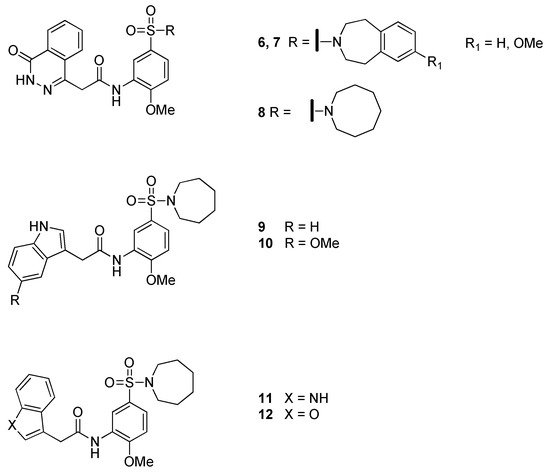
Figure 6. Chemical structure of compounds 6–12.
For additional structural studies, please refer to the article from which this entry is adapted [9].
References
- John D. McKinney; Kerstin Höner Zu Bentrup; Ernesto J. Muñoz-Elías; Andras Miczak; Bing Chen; Wai-Tsing Chan; Dana L Swenson; James C. Sacchettini; William R. Jacobs Jr; David Russell; et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature 2000, 406, 735-738, 10.1038/35021074.
- Sacchettini, J.C.; McKinney, J.D.; Russell, D.G.; Jacobs, W.R., Jr.; Sharma, V.; Sharma, S.; Hener zu Bentrup, K. Isocitrate Lyase Enzyme from Mycobacterium tuberculosis and Inhibitory Agents to Combat Persistent Infection. US Patent WO2002033118, 30 May 2003.
- Sunghark Kwon; Hye Lin Chun; Hyun Ji Ha; So Yeon Lee; Hyun Ho Park; Heterogeneous multimeric structure of isocitrate lyase in complex with succinate and itaconate provides novel insights into its inhibitory mechanism. PLOS ONE 2021, 16, e0251067, 10.1371/journal.pone.0251067.
- Brooke X. C. Kwai; Annabelle J. Collins; Martin J. Middleditch; Jonathan Sperry; Ghader Bashiri; Ivanhoe K. H. Leung; Itaconate is a covalent inhibitor of the Mycobacterium tuberculosis isocitrate lyase. RSC Medicinal Chemistry 2020, 12, 57-61, 10.1039/d0md00301h.
- Markus Ruetz; Gregory C. Campanello; Meredith Purchal; Hongying Shen; Liam McDevitt; Harsha Gouda; Shoko Wakabayashi; Junhao Zhu; Eric J. Rubin; Kurt Warncke; et al.Vamsi K. MoothaMarkos KoutmosRuma Banerjee Itaconyl-CoA forms a stable biradical in methylmalonyl-CoA mutase and derails its activity and repair. Science 2019, 366, 589-593, 10.1126/science.aay0934.
- Moses Barron; PERNICIOUS ANEMIA AND TUBERCULOSIS: IS THERE AN ANTAGONISM?. Journal of the American Medical Association 1933, 100, 1590, 10.1001/jama.1933.02740200024007.
- Monica Kasbekar; Gerhard Fischer; Bryan Mott; Adam Yasgar; Marko Hyvönen; Helena I. M. Boshoff; Chris Abell; Clifton E. Barry; Craig J. Thomas; Selective small molecule inhibitor of the Mycobacterium tuberculosis fumarate hydratase reveals an allosteric regulatory site. Proceedings of the National Academy of Sciences 2016, 113, 7503-7508, 10.1073/pnas.1600630113.
- Andrew J. Whitehouse; M. Daben J. Libardo; Monica Kasbekar; Paul Brear; Gerhard Fischer; Craig J. Thomas; Clifton E Barry; Helena I. M. Boshoff; Anthony G. Coyne; Chris Abell; et al. Targeting of Fumarate Hydratase from Mycobacterium tuberculosis Using Allosteric Inhibitors with a Dimeric-Binding Mode. Journal of Medicinal Chemistry 2019, 62, 10586-10604, 10.1021/acs.jmedchem.9b01203.
- Matteo Mori; Stefania Villa; Samuele Ciceri; Diego Colombo; Patrizia Ferraboschi; Fiorella Meneghetti; An Outline of the Latest Crystallographic Studies on Inhibitor-Enzyme Complexes for the Design and Development of New Therapeutics against Tuberculosis. Molecules 2021, 26, 7082, 10.3390/molecules26237082.
More
Information
Subjects:
Crystallography; Chemistry, Medicinal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
903
Revision:
1 time
(View History)
Update Date:
23 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No