 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simona Bungău | + 3778 word(s) | 3778 | 2020-08-31 05:33:50 | | | |
| 2 | Bruce Ren | Meta information modification | 3778 | 2020-08-31 08:18:08 | | | | |
| 3 | Bruce Ren | + 2 word(s) | 3780 | 2020-09-01 11:02:33 | | | | |
| 4 | Bruce Ren | + 2 word(s) | 3780 | 2020-09-01 11:05:51 | | |
Video Upload Options
This review focuses on what endocannabinoid means related to its possible action in the treatment of Parkinson's disease symptoms, also presenting the role of cannabinoid receptors in these symptoms, but also in terms of the cause and treatment of this pathology.
Current pharmacotherapy of Parkinson’s disease (PD) is symptomatic and palliative, with levodopa/carbidopa therapy remaining the prime treatment, and nevertheless, being unable to modulate the progression of the neurodegeneration. No available treatment for PD can enhance the patient’s life-quality by regressing this diseased state. Various studies have encouraged the enrichment of treatment possibilities by discovering the association of the effects of the endocannabinoid system (ECS) in PD. These reviews delineate the reported evidence from the literature on the neuromodulatory role of the endocannabinoid system and expression of cannabinoid receptors in symptomatology, cause, and treatment of PD progression, wherein cannabinoid (CB) signalling experiences alterations of biphasic pattern during PD progression. Published papers to date were searched via MEDLINE, PubMed, etc., using specific key words in the topic of our manuscript. Endocannabinoids regulate the basal ganglia neuronal circuit pathways, synaptic plasticity, and motor functions via communication with dopaminergic, glutamatergic, and GABAergic signalling systems bidirectionally in PD. Further, gripping preclinical and clinical studies demonstrate the context regarding the cannabinoid compounds, which is supported by various evidence (neuroprotection, suppression of excitotoxicity, oxidative stress, glial activation, and additional benefits) provided by cannabinoid-like compounds (much research addresses the direct regulation of cannabinoids with dopamine transmission and other signalling pathways in PD). More data related to endocannabinoids efficacy, safety, and pharmacokinetic profiles need to be explored, providing better insights into their potential to ameliorate or even regress PD.
1. Introduction
Endocannabinoids, also known as endogenous cannabinoids (eCBs), are the endogenous lipid-based retrograde neurotransmitters that comprise of cannabinoid receptors and imitate the psychomotor effect of Cannabis sativa [1][2][3]. The endocannabinoid system (ECS) is widely known for its ability to regulate numerous physiological roles (modulation of immune system, cognition, appetite regulation, motor function, and pain). In recent times, a variety of studies have been investigating the function of cannabinoids (CBs) in several pathological conditions, including the neurological diseases [4][5][6].
The considerable rise in the scientific interest of promising therapeutic benefits of cannabinoids have displayed a remarkable improvement in parkinsonian symptoms like dyskinesia or tremors [7][8]. Moreover, it has led to an increasing interest in research exploring the potential of these CBs and ECS as possible medical interventions for the treatment of various diseases, including neurodegenerative disorders [8]. The involvement of the cannabinoid 1 (CB1) receptor in ECS is a well expressed phenomenon in basal ganglia, which is reported to be affected in case of different motor dysfunctions and other neurodegenerative disorders including PD [9]. Additionally, the role of ECS in basal ganglia functioning and cortico-striatal pathway regulation has been explored competently in PD models [10][11][12][13]. PD is known as a progressive, chronic neurodegenerative disorder, which has become a considerable social and medical concern, while becoming a financial burden on public health systems in many countries.
The dopaminergic neuronal loss and α-synuclein aggregation in the Lewy bodies that invades the substantia nigra pars compacta (SNpc) are the major pathognomonic hallmarks of this disease [14][15]. Considered to be a multiplex of various environmental, genetic, and age-related factors, the aetiology of PD is still a widely disputed topic [16]. Although well-marked environmental or genetic factors tend to be correlated by several cases, the combination of unknown and unspecified genetic and environmental processes accounted for the majority of cases [17][18]. The deterioration of motor functions is easy cognoscible because of the following typical clinical observable symptoms in PD patients: rigidity, akinesia, bradykinesia, hypokinesia, stooped posture, postural instability, rest tremors, etc., all of them often existing. Cognitive impairment, olfactory dysfunction, psychiatric symptoms, and autonomic dysfunction are common non-motor features. Such motor functions are used to track the therapy response and assess improvement in PD patients [19][20][21]. However, levodopa remains the main symptomatic therapy of PD. Its persistent use in the initial years of the therapy is associated with motor fluctuations in 62% of patients and levodopa-induced dyskinesia (LID) that affects 91% of the patients, as reported by a 10-year prospective study in PD patients [22][23].
Thus, the ongoing study aims at employing newer non-dopaminergic substances that can prevent LID and alleviate motor symptoms. One such engrossing group of agents is represented by the CBs that have not only revealed their neuroprotective capability but also their potential to relieve motor and non-motor symptoms as detected in patients with PD, in various preclinical and clinical studies [23][24][25][26]. However, there is a shortfall of clinical data on the utilization of CBs in patients with PD but preclinical findings indicate that the modulation of the CB signalling pathway could incapacitate and improve motor impairments and symptoms [27][28][29]. The present review aims to lay out an overview of endocannabinoid and its probable influence for the treatment of parkinsonian symptoms, as well as for the expression of cannabinoid receptors in the symptomatology, cause, and treatment of PD progression (wherein cannabinoid signalling experiences alterations in the biphasic pattern and biochemical interactions between CBs, dopamine (DA), and ECS-targeted therapeutic interventions). Published literature data in the field were searched via the most well-known medical data bases (MEDLINE, PubMed, etc.), using specific key words in the topic of our manuscript, and resulting in 202 references mentioned at the final of this review type article.
2. The Endocannabinoid System
Notable advancement in our comprehension of the ECS had been established from the last 15 years. The ECS is a biological lipid-transmitting cascade that includes eCBs, molecules derived from arachidonic acid, and membrane phospholipids. The activity of CBs is expressed via Gi/o protein-coupled receptors, the CB1 receptor, and the cannabinoid 2 (CB2) receptor, being mediated via binding of the ligands to the metabotropic receptor; as well, other receptors are mentioned below, along with the enzymes and agents responsible for the synthesis, degradation, and transportation of eCB, which are essential elements of the body in both physiological and pathological aspects [30][31][32]. The detailed understanding of cannabinoid receptors, their specific locations and actions in the brain was followed by isolation of 2-endogenous arachidonic acid-derived ligands, anandamide/arachidonoyl ethanol amide (AEA), and 2-arachidonoyl glycerol (2-AG), the best-identified eCBs where the former was first to be isolated from porcine brain, and the latter obtained from canine intestines [33][34][35][36][37][38][39].
2.1. Cannabinoid Receptors (CB1 and CB2 Receptors) and Other ECS Associated Receptors
Cannabinoid 1 receptors (CB1 receptors) are crucial signalling mediators predominantly found in the peripheral and central neurons and are best characterized on both the gamma-aminobutyric acid (GABA) and glutamatergic neurons, possessing an excitatory or inhibitory activity. They are enabled in response to the depolarization regulating functions such as pain perception, memory, etc. [40][41][42][43]. Except for conventional neurotransmitters, eCBs are not retained in the synaptic vesicles; rather they act on presynaptic receptors in a retrograde manner after their synthesis and release on demand. It is assumed to be an essential aspect of the neuronal CB1 receptor element that modulates the neurotransmitter release, further establishing homeostasis by avoiding an extreme neuronal activity developing in the central nervous system (CNS) [44]. Higher levels of CB1 receptor are observed in the globus pallidus (GP), the molecular layer of the cerebellum, substantia nigra (SN), hippocampus, and caudate putamen . The GP and SN (two protruding regions engaged in movement control) not only have the strongest CB1 receptor concentrations but rather the strongest eCB concentrations, specifically the N-arachidonoyl-ethanol amine (AEA) [45][46]. The CNS and supraspinal areas of the brain are extensively expressed with one of the primary receptors of CB compounds, i.e., the CB1 receptor involved in nociceptive transmission in the brain. The CB1 receptor is also found to be expressed in the frontal-limbic regions of the brain, which significantly regulate the emotional manifestations of the human brain [47]. CB1 receptor regulates a descending inhibitory pathway from the supraspinal level to the spinal cord nociceptive system via inhibiting GABA release [48][49]. At the spinal cord level, the CB1 receptor significantly mediates the noxious stimulation to the brain [50]. Furthermore, the CB1 receptor is also responsible for confining pain signal propagation, contributing to the peripheral analgesic effects [51].
The expression of CB2 receptors is primarily observed in the immune system such as macrophages, lymphoid organs, T and B cells, and immunocompetent cells at the peripheral regions and modifies numerous aspects such as lymphocytes proliferation, development of cytokines, and cell-mediated immune reactions. Indeed, scientific findings have shown that they often exist in CNS but at rates lower than the CB1 receptor. CB2 receptors situated on astrocytes and microglia, do not seem to play a major role in the cortico-striato-pallidal-circuit modulation. Instead, these are primarily upregulated in response to cytotoxic and neuroinflammatory injury [52]. However, the CB1 receptor is abundantly expressed in the basal ganglia and is actually denser in the substantia nigra (SN), indicating a probable role in the motor control movement [53][54]. Though, these nigrostriatal neurons do not express CB1 receptor themselves, they do express transient receptor-potential type-1 vanilloid (TRPV1) receptors that are activated by AEA [55].
Cannabinoid 2 receptor (CB2 receptor) activation mainly regulates microglial stimulation and recruitment and therefore plays a significant part in neuroinflammation, along with the inflammatory reaction identified in neurological disorders as demonstrated in animal models [56][57][58]. It could be specifically critical in controlling astrocyte action caused by various cytotoxic insults that could solely be caused by CB2 receptor stimulation or may include the CB1 receptor, individually or collectively with the CB2 receptor. Regardless of the type of CB receptor-associated, the advantages that are delivered seem to be correlated mostly with the trophic function performed by the glial cells (enhancements within the distribution of metabolic substrates to neurons). They may indeed promote the levels of anti-inflammatory mediators (transforming growth factor-β as pro-survival; TGF-β), interleukin (IL)-1 receptor antagonists, IL-10, or neurotropin production that could retrieve impaired neurons [59]. The CB2 receptor activation by microglial cells diminishes the generation of neurotoxic elements (i.e., tumour necrosis factor-α (TNF-α)), which is a key player in the pathology involved in brain injury. Activation of CB2 receptors restricts TNF-α development by suppressing nuclear factor-kappa B protein (NF-κB), a transcription factor that plays a vital role in monitoring the inflammatory reaction [60]. The latest evidence revealed the involvement of the CB2 receptor in some neural subpopulations in the brain, endorsing the potential role of the receptor in synaptic processing, though it has still not been established comprehensively. There is no confirmation about the stimulation of such neuronal CB2 receptor showing the neuroprotective effect, but they can appear as a reliable biomarker for neuronal dysfunction in neurodegenerative conditions such as PD [61][62][63][64][65].
Another receptor implicated in the movement control is a protein named transient receptor potential cation channel subfamily V member 1 (capsaicin receptor and vanilloid receptor 1, encoded TRPV1 gene), found in the dopaminergic neurons mainly basal ganglia circuit and sensory neurons. Endo-vanilloid-nociceptive stimuli particularly activate the molecular integrators, namely the TRPV1 receptors. TRPV1 communicates with the eCB, wherein AEA is one of the principal endogenous activators. Studies have also shown that the stimulation of vanilloid receptors can inhibit motor activity, indicating that TRPV1 receptors may perform a role in regulating motor function [66][67][68][69].
G protein-coupled receptor 55 (GPR55), an orphan G-protein receptor expressed widely in the brain (mainly in the striatum), is also a promising target in the PD therapy as it is thought to be involved in the motor function [70][71]. Even though, it lacks a classic CB binding pocket, GPR55 was regarded as “the 3rd CB receptor” and its signalling can be affected and activated by CBs [72][73][74]. In the PD mouse model, downregulation of the GPR55 expression was reported in the striatum and treatment with the abnormal-cannabidiol (Ab-CBD, a synthetic cannabidiol (CBD) isomer and GPR55 agonist) has shown a neuroprotective effect on dopaminergic neuronal cells and improved the motor behaviour [75]. An anatomical, molecular, and behavioural analysis of GPR55−/− mice models showed normal brain structure development and did not influenced ECS and motor learning. The mice displayed flaws in motor coordination, suggesting the involvement of GPR55 signalling in neurodegeneration and modulation of cytokines [76]. The symptomatic impact of Abn-CBD and two other GPR55 agonists (CID1792197 and CID2440433) was studied using the catalepsy test, which indicated GPR55 as a probable symptomatic target for PD .
2.2. Dopamine and Cannabinoid Interactions
Throughout the areas of the brain mentioned above that are significant for many of these neuropsychiatric disorders, CB-1R stimulation could render any of the following:
-
Directly interacts and regulates the expression of dopaminergic neurons (by forming heteromers with dopaminergic receptors, as shown in Figure 1).
-
Impedes the transduction of dopamine (DA) signals in CB-co-localized postsynaptic DA receptors.
CB1 receptors are not located on dopaminergic cells but influence the output of DA by modulating the release of neurotransmitters from projecting excitatory and inhibitory terminals via CB1 receptor stimulation [77][78]. As revealed by additional evidence, the existence of the CB2 receptor in nigrostriatal dopaminergic cells may permit the direct dopaminergic activity control via the endogenous CB mechanism (i.e., the random and elicited discharge, production, secretion of DA, etc.) [79][80]. ECS stimulation has been correlated with motor suppression and declined dopaminergic function (shown in Figure 1).
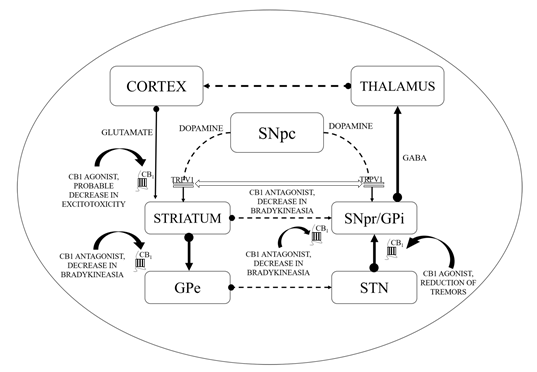
Figure 1. Basal ganglia circuitry and mechanisms depicting the cannabinoid (CB) targets in motor disability improvement in Parkinson’s disease (PD). CB1—Cannabinoid 1 receptor; GABA—γ-aminobutyric acid; GPe—Globus pallidus (external); GPi—Globus pallidus (internal); SNpc—Substantia nigra pars compacta; SNpr—Substantia nigra pars reticulata; STN—subthalamic nucleus; TRPV1—Transient receptor potential type-1 vanniloid; dotted line arrows—underactive; full line arrows—overactive.
The heteromer of the CB1–dopamine 2 receptor (D2) creates a better example of G-protein switching within the heteromer. Thus, CB1 and D2 receptors are generally coupled with Gi/o proteins. Co-stimulation of CB1–D2 receptors in these cellular models leads to a protein-dependent activation of adenylyl-cyclase by Gs [81][82]. Several other researches described simple co-expression of D2 and CB1 receptors is likely to elicit the stimulation of adenylyl cyclase in response to CB1 receptor activation [83]. In various elements of striatal spine modules, D2 and CB1 receptor are colocalized. As stated above, D2 receptors are widely expressed in GABAergic dendritic spines. In addition, D2 receptors are also identified to be widely distributed in dopaminergic, GABAergic, and glutamatergic terminals in the striatal spine modules, wherein their stimulation suppresses the release of neurotransmitters [84][85][86]. Hence the probable localization of CB1–D2 receptor heteromeric may occur both pre- and post-synaptic (i.e., GABAergic enkephalinergic neuron dendritic spines) [87]. Experimental studies in rat striatal membrane provided extra strong evidence, demonstrating an intramembrane receptor–receptor interaction. CB1 receptor stimulation in these preparations declined D2 receptor affinity to DA 88][89][90]. These two features of the CB1–D2 receptor heteromeric, the antagonistic intramembrane interaction and G protein switching, would assume that activation of the CB1 receptor in functional experimental studies must antagonize the effects of striatal neurotransmission mediated by the D2 receptor. It is well established that agonists and antagonists of the CB1 receptor prevent and potentiate motor-mediated effects of D2 receptors, respectively [91][92][93]. Thus, it was proposed that the agonist-mediated motor-depressant effects of the CB1 receptor rely on their ability to prevent the neurotransmission of DA. Since motor activation induced by D2 receptor agonists is mostly dependent on postsynaptic D2 receptors, the behavioural studies strongly suggest that striatal CB1–D2 heteromers are localized in the dendritic spines of GABAergic enkephalinergic neurons. As indicated by this study, another locus where CB1–D2 heteromerisation could partially explain the CB1–D2 receptor interactions at the behavioural level are the terminals of the GABAergic enkephalinergic neurons in the GP [94].
Conventionally, limited eCB tone in hyperkinetic situations accompanies greater dopaminergic activity and the reversible pattern is noticed in hypokinetic motion disorders [95]. CBs could enhance the hypokinetic influence of DA-depleting agents in experimental PD models and lessen the incidence of drugs that produce DA receptor hyperstimulation [96]. Interaction between CB and DA appears to be much more complicated at the cellular level. In the striatum, the levels of eCBs are affected by the dopaminergic transmission initially as depicted by the higher levels of AEA following the stimulation of the D2 receptors as shown in Figure 1 [97]. Such phenomenon is based on either the suppression of its inhibition or the stimulation of its synthesis as proposed by the ability of the D2 receptor agonists to further modify the activity of the fatty acid amide hydrolase (FAAH) and N-acyl-phosphatidylethanolamine (NAPE) phospholipase D. Such activity of CBs stimulated by DA counteracts the action of D2 receptor in the striatum, indicating an inhibitory process focused at restricting the hyperkinetic effect of DA. To introduce extra entanglements, the observations that AEA generated by DA stimulation may intensify the effectiveness of D2 receptor activation, are also indicating cooperative behaviour of CB1 and D2 receptors [10,98–100]. However, suppression of GABA transmission through D2 receptors may slightly be avoided by the CB receptor blockade, indicating eCBs to serve as downward effectors for D2 receptors. While both CB1 and D2 receptors are presented on striatum at the GABA terminals, the intricate relationship between DA and eCBs clearly defines the reconfiguration of such systems in both experimental and idiopathic PD [97]. Earlier studies showed improved eCB behaviour in the basal ganglia of experimental PD along with increased levels of CB1 receptors, AEA, CB1 messenger ribonucleic acid (mRNA), and reduced CB clearance. Similarly, the cerebrospinal fluid of untreated PD patients was found to possess elevated rates of AEA. In the basal ganglia, higher expression of CB1 receptor has indeed been reported. Such alterations are linked with movement inhibition and might even be altered by persistent levodopa therapy. While some of these modifications that represent endogenous compensatory processes have been pointed to minimize the impact of DA failure in the basal ganglia, others would be likely to lead to the progression of typical motor symptoms of PD [97][98][99][100].
3. Alterations Observed in ECS and Basal Ganglia in PD
The significant function of the ECS in PD is shown by the latest shreds of evidence gathered from various studies. The ECS elements are distributed strongly in the basal ganglia neural circuit (as presented in Figure 1) that is part of a dynamic neuronal network that connects bidirectionally with the glutamatergic, GABAergic, and dopaminergic signalling processes in the basal ganglia (Figure 2) [101].
Cannabinoids (CBs) play a prominent part in regulating communication of the striatal and cortical neuronal synapses, controlling the activation of a specific type of synaptic plasticity and altering motor functions. The dopaminergic neuronal cell depletion occurs in PD, mainly results in reduced DA rates in striatum that leads to the alterations in the equilibrium between both the directly and indirectly acting eCB expression and basal ganglia pathways [102]. The foregoing cannabinoid signalling mechanism exhibits a biphasic transition pattern during PD progression [103]. Initially, presymptomatic and early stages of PD are marked by neuronal deterioration along with very little proof of cell death of neurons, which are correlated with CB1 receptor desensitization and exacerbation of cytotoxic damage (i.e., oxidative stress, excitotoxicity, and glial activation) [104]. The advanced/later and intermediate phases of PD are marked by extreme nigral deterioration and severe symptoms of PD, along with upregulated CB1 receptor activities and eCB ligands [104].
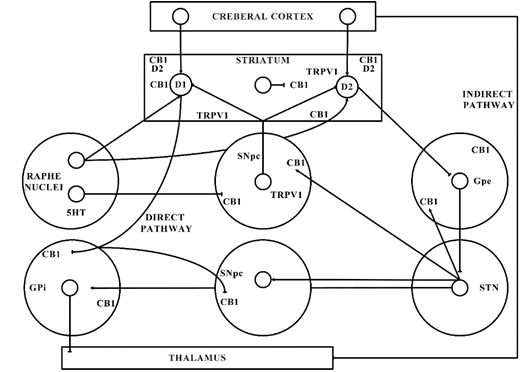
Figure 2. Basal ganglia organization. CB1—Cannabinoid 1 receptor; D1—Dopamine 1 receptor; D2—Dopamine 2 receptor; GABA—γ-aminobutyric acid; GPe—Globus pallidus external; GPi—Globus pallidus internal; SNpc—Substantia nigra pars compacta; STN—subthalamic nucleus; TRPV1—Transient receptor potential type-1.
This might elaborate the capability of ligands with CB receptors to alleviate common PD symptoms. GABAergic neurons engages the internally and externally located parts of the SN and GP within the projecting GABAergic neurons of the brain, also regarded as medium spiny neurons (MSNs) that project to the nuclei of the basal ganglia and provide the striatal output via CB1 receptor expression[105][106]. The CB1 receptor is found in immune-reactive parvalbumin, interneurons, nitric oxide synthase (NOS) neurons, and cholinergic interneurons in the striatum [107]. On presynaptic axons, eCBs stimulate the CB1 receptor to diminish the release of neurotransmitters and glutamates and to serve as synaptic retrogressive messengers produced from post-synaptic neurons. In the same way, the CB1 receptor activation restrains both the release of GABA from striatal afferents and glutamate from cortex and thalamus . CB1 presynaptic receptor activation in the outer parts of the GP may elevate the amount of GABA by minimizing its reuptake to the nucleus from striatal afferents and by lowering its release from striatal afferents in the SN as shown in Figure 2. Depending on all of these pieces of evidence, the activity of the neuronal basal ganglia system is considered to be regulated by the eCBs. The involvement of eCB signalling processes and their association with glutamatergic, GABAergic, and dopaminergic neurotransmitter signalling routes in various neuronal structures renders the ECS as a suitable prospective for a novel PD therapy .
4. Consequences of Basal Ganglia and Cortico-Striatal Plasticity in PD Associated Long Term Depression (LTD)
It is well established that synapses serve durable morphological and functional modulations throughout the basal ganglia neural circuit, especially in the striatum following the continuous stimulation of neuronal mechanisms. This interesting ability regarded as “synaptic plasticity” is assumed to take place at the levels of neuronal circuit dynamics and development, cortico-striatal synapses, and various key cognitive processes, mainly motor associated learning features . DA plays a crucial role in creating two opposing kinds of synaptic cortico-striatal plasticity: long-term depression (LTD) and long-term potentiation (LTP). LTD renders glutamatergic synapses quite less excitable for potential future activation and LTP strengthens the connection between the cortical and striatal neurons. The LTP reversal is called depotentiation (LTP-D) and manages to adjust synaptic signalling to its natural state [108][109]. Although, LTD and LTP-D decrease the strength of synaptic signalling, depotentiation is itself unable to drag down non-potentiated synapses and necessitates the activation of N-methyl-d-aspartate (NMDA) receptors. Prior observations have already revealed that heterosynaptic LTP-D necessitates these receptors (CB1, adenosine A1, GABA-A, p38, mitogen-activated protein kinase (MAPK), and extracellular signal-regulated kinase (ERK) 1/2 signalling), implying that eCBs play a convoluted function in both pre- and post-synaptic changes [110][111][112][113]. In the mesencephalic brain areas, CB1 receptor activation has been shown to elevate the release of acetylcholine, thereby reducing the cholinergic deficit locally in PD [114]. Furthermore, CBs interact with the serotonergic system to influence the LID; loss of nerve supply in striatal dopaminergic renders a levodopa conversion that contribute to a non-physiologic throbbing DA release (false transmitter) [115]. As per one study, 2-AG metabolism takes place through the monoacylglycerol lipase (MAGL), the hydrolysing enzyme in this process. As per another experimental study, serine hydrolase ABHD6 (alpha/beta-hydrolase domain-6) knockdown has shown to decline the hydrolysis of 2-AG, influencing the migration of cells-induced by 2-AG stimulation in vitro [116]. In some studies, alteration in the synaptic plasticity (by genetic ablation of MAGL in mouse cerebellum and hippocampus) is reported to contribute to their desensitization through the mobilization of 2-AG and constant stimulation of CB1 receptor [117][118]. Similarly, the elevation of CB1-dependent LTD occurs by ABHD6 inhibition in mouse cortical excitatory synapses. ABHD6 also regulates the concentration of 2-AG that extends to the presynaptic CB1 receptor [116].
References
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84, doi:10.1016/j.phrs.2009.02.010.
- Di Marzo, V.; Melck, D.; Bisogno, T.; De Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends. Neurosci. 1998, 21, 521–528, doi:10.1016/s0166-2236(98)01283-1.
- Lu, H.C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525, doi:10.1016/j.biopsych.2015.07.028.
- Hanus, L.O.; Mechoulam, R. Novel natural and synthetic ligands of the endocannabinoid system. Curr. Med. Chem. 2010, 17, 1341–1359, doi:10.2174/092986710790980096.
- Maccarrone, M. The Endocannabinoid System and its Manifold Central Actions. In Handbook of Neurochemistry and Molecular Neurobiology: Neural Lipids; Lajtha, A., Tettamanti, G., Goracci, G., Eds.; Springer: Boston, MA, USA, 2009; pp. 385–405, doi:10.1007/978-0-387-30378-9_16.
- Grosu, F.; Ungureanu, A.; Bianchi, E.; Moscu, B.; Coldea, L.; Stupariu, A.L.; Pirici, I.; Roman-Filip, C.C. Multifocal and multicentric low-grade oligoastrocytoma in a young patient. Rom. J. Morphol. Embryol. 2017, 58, 207–210.
- Buhmann, C.; Mainka, T.; Ebersbach, G.; Gandor, F. Evidence for the use of cannabinoids in Parkinson’s disease. J. Neural. Transm. (Vienna) 2019, 126, 913–924, doi:10.1007/s00702-019-02018-8.
- Mainka, T.; Stork, J.; Hidding, U.; Buhmann, C. Cannabis bei Parkinson—Hype oder Heilmittel? [Cannabis in Parkinson’s Disease: Hype or help?]. Fortschr. Neurol. Psychiatr. 2018, 86, 106–116, doi:10.1055/s-0043-120668.
- Abdel-Daim, M.M.; El-Tawil, O.S.; Bungau, S.G.; Atanasov, A.G. Applications of Antioxidants in Metabolic Disorders and Degenerative Diseases: Mechanistic Approach. Oxidative Med. Cell. Longev. 2019, 2019, doi:10.1155/2019/4179676.
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000, 14, 1432–1438, doi:10.1096/fj.14.10.1432.
- Gubellini, P.; Picconi, B.; Bari, M.; Battista, N.; Calabresi, P.; Centonze, D.; Bernardi, G.; Finazzi-Agrò, A.; Maccarrone, M. Experimental parkinsonism alters endocannabinoid degradation: Implications for striatal glutamatergic transmission. J. Neurosci. 2002, 22, 6900–6907, doi:10.1523/JNEUROSCI.22-16-06900.2002.
- Hernandes, M.S.; Café-Mendes, C.C.; Britto, L.R. NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxid. Med. Cell Longev. 2013, 2013, doi:10.1155/2013/157857.
- Pisani, A.; Fezza, F.; Galati, S.; Battista, N.; Napolitano, S.; Finazzi-Agrò, A.; Bernardi, G.; Brusa, L.; Pierantozzi, M.; Stanzione, P.; et al. High endogenous cannabinoid levels in the cerebrospinal fluid of untreated Parkinson’s disease patients. Ann. Neurol. 2005, 57, 777–779, doi:10.1002/ana.20462.
- Radhakrishnan, D.M.; Goyal, V. Parkinson’s disease: A review. Neurol. India 2018, 66, S26–S35, doi:10.4103/0028-3886.226451.
- Zeng, X.S.; Geng, W.S.; Jia, J.J.; Chen, L.; Zhang, P.P. Cellular and Molecular Basis of Neurodegeneration in Parkinson Disease. Front. Aging Neurosci. 2018, 10, 109, doi:10.3389/fnagi.2018.00109.
- Abdel-Daim, M.M.; Zakhary, N.I.; Aleya, L.; Bungau, S.G.; Bohara, R.A.; Siddiqi, N.J. Aging, Metabolic, and Degenerative Disorders: Biomedical Value of Antioxidants. Oxidative Med. Cell. Longev. 2018, doi:10.1155/2018/2098123.
- Kaur, G.; Behl, T.; Bungau, S.; Kumar, A.; Uddin, M.S.; Mehta, V.; Zengin, G.; Mathew, B.; Shah, M.A.; Arora, S. Dysregulation of the Gut-Brain Axis, Dysbiosis and Influence of numerous factors on Gut Microbiota associated Parkinson’s Disease. Curr. Neuropharmacol. 2020, 18, 1–15, doi:10.2174/1570159X18666200606233050.
- Desai Bradaric, B.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural. Transm. (Vienna) 2012, 119, 59–71, doi:10.1007/s00702-011-0684-8.
- Kouli, A.; Torsney, K.M.; Kuan, W.L. Parkinson’s Disease: Etiology, Neuropathology, and Pathogenesis. In Parkinson’s Disease: Pathogenesis and Clinical Aspects [Internet]; Codon Publications: Brisbane, Australia, 2018; doi:10.15586/codonpublications.parkinsonsdisease.2018.ch1.
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85, doi:10.1016/j.arr.2017.12.007.
- Schapira, A.H. Etiology of Parkinson’s disease. Neurology 2006, 66, S10–S23, doi:10.1212/wnl.66.10_suppl_4.s10.
- Gupta, M.; Kant, K.; Sharma, R.; Kumar, A. Evaluation of In Silico Anti-parkinson Potential of β-asarone. Cent. Nerv. Syst. Agents Med. Chem. 2018, 18, 128–135, doi:10.2174/1871524918666180416153742.
- Rascol, O.; Payoux, P.; Ory, F.; Ferreira, J.J.; Brefel-Courbon, C.; Montastruc, J.L. Limitations of current Parkinson’s disease therapy. Ann. Neurol. 2003, 53, S3–S15, doi:10.1002/ana.10513.
- Utsumi, H.; Okuma, Y.; Kano, O.; Suzuki, Y.; Iijima, M.; Tomimitsu, H.; Hashida, H.; Kubo, S.; Suzuki, M.; Nanri, K.; et al. Evaluation of the efficacy of pramipexole for treating levodopa-induced dyskinesia in patients with Parkinson’s disease. Intern. Med. 2013, 52, 325–332, doi:10.2169/internalmedicine.52.8333.
- Fernández-Ruiz, J.; Moreno-Martet, M.; Rodríguez-Cueto, C.; Palomo-Garo, C.; Gómez-Cañas, M.; Valdeolivas, S.; Guaza, C.; Romero, J.; Guzmán, M.; Mechoulam, R.; et al. Prospects for cannabinoid therapies in basal ganglia disorders. Br. J. Pharmacol. 2011, 163, 1365–1378, doi:10.1111/j.1476-5381.2011.01365.x.
- Laprairie, R.B.; Bagher, A.M.; Kelly, M.E.M.; Denovan-Wright, E.M. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br. J. Pharmacol. 2015, 172, 4790–4805, doi:10.1111/bph.13250.
- ElSohly, M.A.; Gul, W. Constituents of Cannabis Sativa. In Handbook of Cannabis; Oxford University Press: Oxford, UK, 2014; pp. 3–22, doi:10.1093/acprof:oso/9780199662685.003.0001.
- García, M.C.; Cinquina, V.; Palomo-Garo, C.; Rábano, A.; Fernández-Ruiz, J. Identification of CB2 receptors in human nigral neurons that degenerate in Parkinson’s disease. Neurosci. Lett. 2015, 587, 1–4, doi:10.1016/j.neulet.2014.12.003.
- Kluger, B.; Triolo, P.; Jones, W.; Jankovic, J. The therapeutic potential of cannabinoids for movement disorders. Mov. Disord. 2015, 30, 313–327, doi:10.1002/mds.26142.
- Behl, T.; Kaur, I.; Kotwani, A. Role of endocannabinoids in the progression of diabetic retinopathy. Diabetes Metab. Res. Rev. 2016, 32, 251–259, doi:10.1002/dmrr.2710.
- De Petrocellis, L.; Di Marzo, V. Non-CB1, non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: Focus on G-protein-coupled receptors and transient receptor potential channels. J. Neuroimmune Pharmacol. 2010, 5, 103–121, doi:10.1007/s11481-009-9177-z.
- Gasperi, V.; Fezza, F.; Pasquariello, N.; Bari, M.; Oddi, S.; Agrò, A.F.; Maccarrone, M. Endocannabinoids in adipocytes during differentiation and their role in glucose uptake. Cell Mol. Life Sci. 2007, 64, 219–229, doi:10.1007/s00018-006-6445-4.
- Cascio, M.G.; Marini, P. Biosynthesis and Fate of Endocannabinoids. In Endocannabinoids; Pertwee, R.G., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 39–58, doi:10.1007/978-3-319-20825-1_2.
- Di Marzo, V. Targeting the endocannabinoid system: To enhance or reduce? Nat. Rev. Drug Discov. 2008, 7, 438–455, doi:10.1038/nrd2553.
- Di Marzo, V.; Bisogno, T.; De Petrocellis, L. Anandamide: Some like it hot. Trends Pharmacol. Sci. 2001, 22, 346–349, doi:10.1016/s0165-6147(00)01712-0.
- Hashimotodani, Y.; Ohno-Shosaku, T.; Kano, M. Presynaptic monoacylglycerol lipase activity determines basal endocannabinoid tone and terminates retrograde endocannabinoid signaling in the hippocampus. J. Neurosci. 2007, 27, 1211–1219, doi:10.1523/JNEUROSCI.4159-06.2007.
- Ivanov, I.; Borchert, P.; Hinz, B. A simple method for simultaneous determination of N-arachidonoylethanolamine, N-oleoylethanolamine, N-palmitoylethanolamine and 2-arachidonoylglycerol in human cells. Anal. Bioanal. Chem. 2015, 407, 1781–1787, doi:10.1007/s00216-014-8384-5.
- Liu, J.; Batkai, S.; Pacher, P.; Harvey-White, J.; Wagner, J.A.; Cravatt, B.F.; Gao, B.; Kunos, G. Lipopolysaccharide induces anandamide synthesis in macrophages via CD14/MAPK/phosphoinositide 3-kinase/NF-kappaB independently of platelet-activating factor. J. Biol. Chem. 2003, 278, 45034–45039, doi:10.1074/jbc.M306062200.
- Pacher, P.; Kunos, G. Modulating the endocannabinoid system in human health and disease: Successes and failures. FEBS J. 2013, 280, 1918–1943, doi:10.1111/febs.12260.
- Giuffrida, A.; Martinez, A. The Endocannabinoid System and Parkinson Disease. In The Endocannabinoid System; Murillo-Rodríguez, E., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 63–81, doi:10.1016/B978-0-12-809666-6.00003-4.
- Pertwee, R.G. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br. J. Pharmacol. 2008, 153, 199–215, doi:10.1038/sj.bjp.0707442.
- Wilson, R.I.; Nicoll, R.A. Endocannabinoid signaling in the brain. Science 2002, 296, 678–682, doi:10.1126/science.1063545.
- Xu, J.Y.; Chen, C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 2015, 21, 152–168, doi:10.1177/1073858414524632.
- Hegde, V.L.; Nagarkatti, M.; Nagarkatti, P.S. Cannabinoid receptor activation leads to massive mobilization of myeloid-derived suppressor cells with potent immunosuppressive properties. Eur. J. Immunol. 2010, 40, 3358–3371, doi:10.1002/eji.201040667.
- Di Marzo, V.; Berrendero, F.; Bisogno, T.; González, S.; Cavaliere, P.; Romero, J.; Cebeira, M.; Ramos, J.A.; Fernández-Ruiz, J.J. Enhancement of anandamide formation in the limbic forebrain and reduction of endocannabinoid contents in the striatum of delta9-tetrahydrocannabinol-tolerant rats. J. Neurochem. 2000, 74, 1627–1635, doi:10.1046/j.1471-4159.2000.0741627.x.
- Herkenham, M.; Lynn, A.B.; Little, M.D.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 1990, 87, 1932–1936.
- Toczek, M.; Malinowska, B. Enhanced endocannabinoid tone as a potential target of pharmacotherapy. Life Sci. 2018, 204, 20–45, doi:10.1016/j.lfs.2018.04.054.
- Deutsch, D.G. A Personal Retrospective: Elevating Anandamide (AEA) by Targeting Fatty Acid Amide Hydrolase (FAAH) and the Fatty Acid Binding Proteins (FABPs). Front. Pharmacol. 2016, 7, 370, doi:10.3389/fphar.2016.00370.
- Izzo, A.A.; Deutsch, D.G. Unique pathway for anandamide synthesis and liver regeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 6339–6340, doi:10.1073/pnas.1103566108.
- Ueda, N.; Tsuboi, K.; Uyama, T. Metabolic Enzymes for Endocannabinoids and Endocannabinoid-Like Mediators. In The Endocannabinoidome; Di Marzo, V., Wang, J., Eds.; Academic Press: Boston, MA, USA, 2015; pp. 111–135, doi:10.1016/B978-0-12-420126-2.00008-0.
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391, doi:10.1111/bph.12411.
- Ternianov, A.; Pérez-Ortiz, J.M.; Solesio, M.E.; García-Gutiérrez, M.S.; Ortega-Álvaro, A.; Navarrete, F.; Leiva, C.; Galindo, M.F.; Manzanares, J. Overexpression of CB2 cannabinoid receptors results in neuroprotection against behavioral and neurochemical alterations induced by intracaudate administration of 6-hydroxydopamine. Neurobiol. Aging 2012, 33, 421.e1-16, doi:10.1016/j.neurobiolaging.2010.09.012.
- Mailleux, P.; Vanderhaeghen, J.J. Distribution of neuronal cannabinoid receptor in the adult rat brain: A comparative receptor binding radioautography and in situ hybridization histochemistry. Neuroscience 1992, 48, 655–668, doi:10.1016/0306-4522(92)90409-u.
- Herkenham, M.; Lynn, A.B.; Johnson, M.R.; Melvin, L.S.; de Costa, B.R.; Rice, K.C. Characterization and localization of cannabinoid receptors in rat brain: A quantitative in vitro autoradiographic study. J. Neurosci. 1991, 11, 563–583, doi:10.1523/JNEUROSCI.11-02-00563.1991.
- Mezey, E.; Tóth, Z.E.; Cortright, D.N.; Arzubi, M.K.; Krause, J.E.; Elde, R.; Guo, A.; Blumberg, P.M.; Szallasi, A. Distribution of mRNA for vanilloid receptor subtype 1 (VR1), and VR1-like immunoreactivity, in the central nervous system of the rat and human. Proc. Natl. Acad. Sci. USA 2000, 97, 3655–3660, doi:10.1073/pnas.060496197.
- Carta, A.R.; Pisanu, A.; Carboni, E. Do PPAR-Gamma Agonists Have a Future in Parkinson’s Disease Therapy? Parkinsons. Dis. 2011, 2011, doi:10.4061/2011/689181.
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884, doi:10.1038/nrn1247.
- Price, D.A.; Martinez, A.A.; Seillier, A.; Koek, W.; Acosta, Y.; Fernandez, E.; Strong, J.R.; Lutz, B.; Marsicano, G.; Roberts, J.L.; et al. WIN55,212-2, a cannabinoid receptor agonist, protects against nigrostriatal cell loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Eur. J. Neurosci. 2009, 29, 2177–2186, doi:10.1111/j.1460-9568.2009.06764.x.
- Fernández-Ruiz, J.; García, C.; Sagredo, O.; Gómez-Ruiz, M.; de Lago, E. The endocannabinoid system as a target for the treatment of neuronal damage. Expert Opin. Ther. Targets 2010, 14, 387–404, doi:10.1517/14728221003709792.
- Fernández-Ruiz, J.; Romero, J.; Velasco, G.; Tolón, R.M.; Ramos, J.A.; Guzmán, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45, doi:10.1016/j.tips.2006.11.001.
- Lanciego, J.L.; Barroso-Chinea, P.; Rico, A.J.; Conte-Perales, L.; Callén, L.; Roda, E.; Gómez-Bautista, V.; López, I.P.; Lluis, C.; Labandeira-García, J.L.; et al. Expression of the mRNA coding the cannabinoid receptor 2 in the pallidal complex of Macaca fascicularis. J. Psychopharmacol. 2011, 25, 97–104, doi:10.1177/0269881110367732.
- López-Sendón Moreno, J.L.; García Caldentey, J.; Trigo Cubillo, P.; Ruiz Romero, C.; García Ribas, G.; Alonso Arias, M.A.; García de Yébenes, M.J.; Tolón, R.M.; Galve-Roperh, I.; Sagredo, O.; et al. A double-blind, randomized, cross-over, placebo-controlled, pilot trial with Sativex in Huntington’s disease. J. Neurol. 2016, 263, 1390–1400, doi:10.1007/s00415-016-8145-9.
- Molina-Holgado, F.; Pinteaux, E.; Moore, J.D.; Molina-Holgado, E.; Guaza, C.; Gibson, R.M.; Rothwell, N.J. Endogenous interleukin-1 receptor antagonist mediates anti-inflammatory and neuroprotective actions of cannabinoids in neurons and glia. J. Neurosci. 2003, 23, 6470–6474, doi:10.1523/jneurosci.23-16-06470.2003.
- Rodríguez-Cueto, C.; Benito, C.; Fernández-Ruiz, J.; Romero, J.; Hernández-Gálvez, M.; Gómez-Ruiz, M. Changes in CB1 and CB2 receptors in the post-mortem cerebellum of humans affected by spinocerebellar ataxias. Br. J. Pharmacol. 2014, 171, 1472–1489, doi:10.1111/bph.12283.
- Smith, S.R.; Denhardt, G.; Terminelli, C. The anti-inflammatory activities of cannabinoid receptor ligands in mouse peritonitis models. Eur. J. Pharmacol. 2001, 432, 107–119, doi:10.1016/s0014-2999(01)01477-7.
- Babayeva, M.; Assefa, H.; Basu, P.; Chumki, S.; Loewy, Z. Marijuana Compounds: A Nonconventional Approach to Parkinson’s Disease Therapy. Parkinsons Dis. 2016, 2016, doi:10.1155/2016/1279042.
- Batista, L.A.; Gobira, P.H.; Viana, T.G.; Aguiar, D.C.; Moreira, F.A. Inhibition of endocannabinoid neuronal uptake and hydrolysis as strategies for developing anxiolytic drugs. Behav. Pharmacol. 2014, 25, 425–433, doi:10.1097/FBP.0000000000000073.
- Dos Anjos-Garcia, T.; Ullah, F.; Falconi-Sobrinho, L.L.; Coimbra, N.C. CB 1 cannabinoid receptor-mediated anandamide signalling reduces the defensive behaviour evoked through GABA A receptor blockade in the dorsomedial division of the ventromedial hypothalamus. Neuropharmacology 2017, 113, 156–166, doi:10.1016/j.neuropharm.2016.04.003.
- Palazzo, E.; Rossi, F.; Maione, S. Role of TRPV1 receptors in descending modulation of pain. Mol. Cell Endocrinol. 2008, 286, S79–S83, doi:10.1016/j.mce.2008.01.013.
- Martínez-Pinilla, E.; Aguinaga, D.; Navarro, G.; Rico, A.J.; Oyarzábal, J.; Sánchez-Arias, J.A.; Lanciego, J.L.; Franco, R. Targeting CB 1 and GPR55 Endocannabinoid Receptors as a Potential Neuroprotective Approach for Parkinson’s Disease. Mol. Neurobiol. 2019, 56, 5900–5910, doi:10.1007/s12035-019-1495-4.
- Celorrio, M.; Rojo-Bustamante, E.; Fernández-Suárez, D.; Sáez, E.; Estella-Hermoso de Mendoza, A.; Müller, C.E.; Ramírez, M.J.; Oyarzábal, J.; Franco, R.; Aymerich, M.S. GPR55: A therapeutic target for Parkinson’s disease? Neuropharmacology 2017, 125, 319–332, doi:10.1016/j.neuropharm.2017.08.017.
- Kotsikorou, E.; Madrigal, K.E.; Hurst, D.P.; Sharir, H.; Lynch, D.L.; Heynen-Genel, S.; Milan, L.B.; Chung, T.D.; Seltzman, H.H.; Bai, Y.; et al. Identification of the GPR55 agonist binding site using a novel set of high-potency GPR55 selective ligands. Biochemistry 2011, 50, 5633–5647, doi:10.1021/bi200010k.
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101, doi:10.1038/sj.bjp.0707460.
- Baker, D.; Pryce, G.; Davies, W.L.; Hiley, C.R. In silico patent searching reveals a new cannabinoid receptor. Trends Pharmacol. Sci. 2006, 27, 1–4, doi:10.1016/j.tips.2005.11.003.
- Saliba, S.W.; Jauch, H.; Gargouri, B.; Keil, A.; Hurrle, T.; Volz, N.; Mohr, F.; van der Stelt, M.; Bräse, S.; Fiebich, B.L. Anti-neuroinflammatory effects of GPR55 antagonists in LPS-activated primary microglial cells. J. Neuroinflamm. 2018, 15, 322, doi:10.1186/s12974-018-1362-7.
- Wu, C.S.; Chen, H.; Sun, H.; Zhu, J.; Jew, C.P.; Wager-Miller, J.; Straiker, A.; Spencer, C.; Bradshaw, H.; Mackie, K.; et al. GPR55, a G-Protein Coupled Receptor for Lysophosphatidylinositol, Plays a Role in Motor Coordination. PLoS ONE 2013, 8, e60314, doi:10.1371/journal.pone.0060314.
- Mátyás, F.; Yanovsky, Y.; Mackie, K.; Kelsch, W.; Misgeld, U.; Freund, T.F. Subcellular localization of type 1 cannabinoid receptors in the rat basal ganglia. Neuroscience 2006, 137, 337–361, doi:10.1016/j.neuroscience.2005.09.005.
- Gerdeman, G.; Lovinger, D.M. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J. Neurophysiol. 2001, 85, 468–471, doi:10.1152/jn.2001.85.1.468.
- Rodríguez De Fonseca, F.; Gorriti, M.A.; Bilbao, A.; Escuredo, L.; García-Segura, L.M.; Piomelli, D.; Navarro, M. Role of the endogenous cannabinoid system as a modulator of dopamine transmission: Implications for Parkinson’s disease and schizophrenia. Neurotox. Res. 2001, 3, 23–35, doi:10.1007/BF03033228.
- García, C.; Palomo-Garo, C.; Gómez-Gálvez, Y.; Fernández-Ruiz, J. Cannabinoid–dopamine interactions in the physiology and physiopathology of the basal ganglia. Br. J. Pharmacol. 2016, 173, 2069–2079, doi:10.1111/bph.13215.
- Kearn, C.S.; Blake-Palmer, K.; Daniel, E.; Mackie, K.; Glass, M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: A mechanism for receptor cross-talk? Mol. Pharmacol. 2005, 67, 1697–1704, doi:10.1124/mol.104.006882.
- Glass, M.; Felder, C.C. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: Evidence for a Gs linkage to the CB1 receptor. J. Neurosci. 1997, 17, 5327–5333, doi:10.1523/JNEUROSCI.17-14-05327.1997.
- Jarrahian, A.; Watts, V.J.; Barker, E.L. D2 dopamine receptors modulate Galpha-subunit coupling of the CB1 cannabinoid receptor. J. Pharmacol. Exp. Ther. 2004, 308, 880–886, doi:10.1124/jpet.103.057620.
- Bamford, N.S.; Robinson, S.; Palmiter, R.D.; Joyce, J.A.; Moore, C.; Meshul, C.K. Dopamine modulates release from corticostriatal terminals. J. Neurosci. 2004, 24, 9541–9552, doi:10.1523/jneurosci.2891-04.2004.
- Centonze, D.; Battista, N.; Rossi, S.; Mercuri, N.B.; Finazzi-Agrò, A.; Bernardi, G.; Calabresi, P.; Maccarrone, M. A critical interaction between dopamine D2 receptors and endocannabinoids mediates the effects of cocaine on striatal gabaergic Transmission. Neuropsychopharmacology 2004, 29, 1488–1497, doi:10.1038/sj.npp.1300458.
- Usiello, A.; Baik, J.H.; Rougé-Pont, F.; Picetti, R.; Dierich, A.; LeMeur, M.; Piazza, P.V.; Borrelli, E. Distinct functions of the two isoforms of dopamine D2 receptors. Nature 2000, 408, 199–203, doi:10.1038/35041572.
- Pickel, V.M.; Chan, J.; Kash, T.L.; Rodríguez, J.J.; MacKie, K. Compartment-specific localization of cannabinoid 1 (CB1) and mu-opioid receptors in rat nucleus accumbens. Neuroscience 2004, 127, 101–112, doi:10.1016/j.neuroscience.2004.05.015.
- Agnati, L.F.; Ferré, S.; Lluis, C.; Franco, R.; Fuxe, K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol. Rev. 2003, 55, 509–550, doi:10.1124/pr.55.3.2.
- Ferré, S.; Ciruela, F.; Woods, A.S.; Lluis, C.; Franco, R. Functional relevance of neurotransmitter receptor heteromers in the central nervous system. Trends Neurosci. 2007, 30, 440–446, doi:10.1016/j.tins.2007.07.001.
- Franco, R.; Casadó, V.; Cortés, A.; Ferrada, C.; Mallol, J.; Woods, A.; Lluis, C.; Canela, E.I.; Ferré, S. Basic concepts in G-protein-coupled receptor homo- and heterodimerization. Sci. World J. 2007, 7, 48–57, doi:10.1100/tsw.2007.197.
- Martín, A.B.; Fernandez-Espejo, E.; Ferrer, B.; Gorriti, M.A.; Bilbao, A.; Navarro, M.; Rodriguez de Fonseca, F.; Moratalla, R. Expression and function of CB1 receptor in the rat striatum: Localization and effects on D1 and D2 dopamine receptor-mediated motor behaviors. Neuropsychopharmacology 2008, 33, 1667–1679, doi:10.1038/sj.npp.1301558.
- Maneuf, Y.P.; Crossman, A.R.; Brotchie, J.M. The cannabinoid receptor agonist WIN 55,212-2 reduces D2, but not D1, dopamine receptor-mediated alleviation of akinesia in the reserpine-treated rat model of Parkinson’s disease. Exp. Neurol. 1997, 148, 265–270, doi:10.1006/exnr.1997.6645.
- Andersson, M.; Usiello, A.; Borgkvist, A.; Pozzi, L.; Dominguez, C.; Fienberg, A.A.; Svenningsson, P.; Fredholm, B.B.; Borrelli, E.; Greengard, P.; et al. Cannabinoid action depends on phosphorylation of dopamine- and cAMP-regulated phosphoprotein of 32 kDa at the protein kinase A site in striatal projection neurons. J. Neurosci. 2005, 25, 8432–8438, doi:10.1523/jneurosci.1289-05.2005.
- Ferré, S.; Goldberg, S.R.; Lluis, C.; Franco, R. Looking for the role of cannabinoid receptor heteromers in striatal function. Neuropharmacology 2009, 56, 226–234, doi:10.1016/j.neuropharm.2008.06.076.
- Fernández-Ruiz, J. The endocannabinoid system as a target for the treatment of motor dysfunction. Br. J. Pharmacol. 2009, 156, 1029–1040, doi:10.1111/j.1476-5381.2008.00088.x.
- Fernández-Ruiz, J.; Lastres-Becker, I.; Cabranes, A.; González, S.; Ramos, J.A. Endocannabinoids and basal ganglia functionality. Prostaglandins Leukot. Essent Fatty Acids 2002, 66, 257–267, doi:10.1054/plef.2001.0350.
- Stampanoni Bassi, M.; Sancesario, A.; Morace, R.; Centonze, D.; Iezzi, E. Cannabinoids in Parkinson’s Disease. Cannabis Cannabinoid Res. 2017, 2, 21–29, doi:10.1089/can.2017.0002.
- Alonso, R.; Voutsinos, B.; Fournier, M.; Labie, C.; Steinberg, R.; Souilhac, J.; Le Fur, G.; Soubrié, P. Blockade of cannabinoid receptors by SR141716 selectively increases Fos expression in rat mesocorticolimbic areas via reduced dopamine D2 function. Neuroscience 1999, 91, 607–620, doi:10.1016/s0306-4522(98)00675-7.
- Meschler, J.P.; Conley, T.J.; Howlett, A.C. Cannabinoid and dopamine interaction in rodent brain: Effects on locomotor activity. Pharmacol. Biochem. Behav. 2000, 67, 567–573, doi:10.1016/s0091-3057(00)00390-7.
- Nava, F.; Carta, G.; Battasi, A.M.; Gessa, G.L. D2 dopamine receptors enable Δ9-tetrahydrocannabinol induced memory impairment and reduction of hippocampal extracellular acetylcholine concentration. Br. J. Pharmacol. 2000, 130, 1201–1210, doi:10.1038/sj.bjp.0703413.
- Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G.; Costa, B. Beneficial effects of a Cannabis sativa extract treatment on diabetes-induced neuropathy and oxidative stress. Phytother. Res. 2009, 23, 1678–1684, doi:10.1002/ptr.2806.
- Bezard, E.; Brotchie, J.M.; Gross, C.E. Pathophysiology of levodopa-induced dyskinesia: Potential for new therapies. Nat. Rev. Neurosci. 2001, 2, 577–588, doi:10.1038/35086062.
- Reddy, V.; Grogan, D.; Ahluwalia, M.; Salles, É.L.; Ahluwalia, P.; Khodadadi, H.; Alverson, K.; Nguyen, A.; Raju, S.P.; Gaur, P.; et al. Targeting the endocannabinoid system: A predictive, preventive, and personalized medicine-directed approach to the management of brain pathologies. EPMA J. 2020, 11, 217–250, doi:10.1007/s13167-020-00203-4.
- García-Arencibia, M.; García, C.; Fernández-Ruiz, J. Cannabinoids and Parkinson’s disease. CNS Neurol Disord. Drug Targets 2009, 8, 432–439, doi:10.2174/187152709789824642.
- DeLong, M.R.; Wichmann, T. Circuits and circuit disorders of the basal ganglia. Arch. Neurol. 2007, 64, 20–24, doi:10.1001/archneur.64.1.20.
- Kawaguchi, Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J. Neurosci. 1993, 13, 4908–4923, doi:10.1523/JNEUROSCI.13-11-04908.1993.
- Calabresi, P.; Picconi, B.; Tozzi, A.; Di Filippo, M. Dopamine-mediated regulation of corticostriatal synaptic plasticity. Trends Neurosci. 2007, 30, 211–219, doi:10.1016/j.tins.2007.03.001.
- Huang, C.C.; Hsu, K.S. Progress in understanding the factors regulating reversibility of long-term potentiation. Rev. Neurosci. 2001, 12, 51–68, doi:10.1515/revneuro.2001.12.1.51.
- Raymond, C.R. LTP forms 1, 2 and 3: Different mechanisms for the “long” in long-term potentiation. Trends Neurosci. 2007, 30, 167–175, doi:10.1016/j.tins.2007.01.007.
- Centonze, D.; Bernardi, G.; Koch, G. Mechanisms of disease: Basic-research-driven investigations in humans—The case of hyperkinetic disorders. Nat. Clin. Pract. Neurol. 2007, 3, 572–580, doi:10.1038/ncpneuro0617.
- Chen, Z.; Xiong, C.; Pancyr, C.; Stockwell, J.; Walz, W.; Cayabyab, F.S. Prolonged adenosine A1 receptor activation in hypoxia and pial vessel disruption focal cortical ischemia facilitates clathrin-mediated AMPA receptor endocytosis and long-lasting synaptic inhibition in rat hippocampal CA3-CA1 synapses: Differential regulation of GluA2 and GluA1 subunits by p38 MAPK and JNK. J. Neurosci. 2014, 34, 9621–9643, doi:10.1523/jneurosci.3991-13.2014.
- Chung, H.J.; Ge, W.P.; Qian, X.; Wiser, O.; Jan, Y.N.; Jan, L.Y. G protein-activated inwardly rectifying potassium channels mediate depotentiation of long-term potentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 635–640, doi:10.1073/pnas.0811685106.
- Izumi, Y.; Zorumski, C.F. GABA and Endocannabinoids Mediate Depotentiation of Schaffer Collateral Synapses Induced by Stimulation of Temperoammonic Inputs. PLoS ONE 2016, 11, e0149034, doi:10.1371/journal.pone.0149034.
- Chagas, M.H.; Eckeli, A.L.; Zuardi, A.W.; Pena-Pereira, M.A.; Sobreira-Neto, M.A.; Sobreira, E.T.; Camilo, M.R.; Bergamaschi, M.M.; Schenck, C.H.; Hallak, J.E.; et al. Cannabidiol can improve complex sleep-related behaviours associated with rapid eye movement sleep behaviour disorder in Parkinson’s disease patients: A case series. J. Clin. Pharm. Ther. 2014, 39, 564–566, doi:10.1111/jcpt.12179.
- Espay, A.J.; Morgante, F.; Merola, A.; Fasano, A.; Marsili, L.; Fox, S.H.; Bezard, E.; Picconi, B.; Calabresi, P.; Lang, A.E. Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann. Neurol. 2018, 84, 797–811, doi:10.1002/ana.25364.
- Marrs, W.R.; Blankman, J.L.; Horne, E.A.; Thomazeau, A.; Lin, Y.H.; Coy, J.; Bodor, A.L.; Muccioli, G.G.; Hu, S.S.J.; Woodruff, G.; et al. The serine hydrolase ABHD6 controls the accumulation and efficacy of 2-AG at cannabinoid receptors. Nat. Neurosci. 2010, 13, 951–957, doi:10.1038/nn.2601.
- Pan, J.P.; Zhang, H.Q.; Wei-Wang.; Guo, Y.F.; Na-Xiao.; Cao, X.H.; Liu, L.J. Some subtypes of endocannabinoid/endovanilloid receptors mediate docosahexaenoic acid-induced enhanced spatial memory in rats. Brain Res. 2011, 1412, 18–27, doi:10.1016/j.brainres.2011.07.015.
- Zhong, P.; Pan, B.; Gao, X.; Blankman, J.L.; Cravatt, B.F.; Liu, Q. Genetic deletion of monoacylglycerol lipase alters endocannabinoid-mediated retrograde synaptic depression in the cerebellum. J. Physiol. 2011, 589, 4847–4855, doi:10.1113/jphysiol.2011.215509.

