Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marcello Miceli | + 3275 word(s) | 3275 | 2022-01-11 09:31:16 | | | |
| 2 | Beatrix Zheng | Meta information modification | 3275 | 2022-01-20 10:12:24 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Miceli, M. ALS2-Related Motor Neuron Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/18540 (accessed on 27 July 2026).
Miceli M. ALS2-Related Motor Neuron Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/18540. Accessed July 27, 2026.
Miceli, Marcello. "ALS2-Related Motor Neuron Diseases" Encyclopedia, https://encyclopedia.pub/entry/18540 (accessed July 27, 2026).
Miceli, M. (2022, January 20). ALS2-Related Motor Neuron Diseases. In Encyclopedia. https://encyclopedia.pub/entry/18540
Miceli, Marcello. "ALS2-Related Motor Neuron Diseases." Encyclopedia. Web. 20 January, 2022.
Copy Citation
Mutations of the ALS2 gene, which encodes for the protein Alsin, are linked to three recessive motor neuron diseases characterized by early onset. Alsin is an intriguing protein characterized by several structured domains with distinct functions.
Alsin
IAHSP
JPLS
JALS
rare diseases
protein
mutations
neurodegenerative
1. Microscopic Level: Molecular Features of IAHSP and Other Related ALS2 Pathologies
The clinical course of the ALS2-induced MND forms has highlighted the need to investigate the molecular basis underlying these pathologies. At the lowest scale of investigation, attention is focused on the ALS2 gene, located on the long arm of chromosome 2q33 and composed of 34 exons. It is transcribed into two spliced transcripts, a long form with 1657 amino acids and a short form with 396 amino acids (Figure 1). Both proteins are expressed in various human tissues, but mostly in the brain (cerebellum) and spinal cord. However, some ectopic expression has been detected in the testis [1]. The larger transcript encodes for a protein called Alsin (molecular weight is 184 kD), localized on the cell cytoplasm, onto the cytoplasmic face of perinuclear and enlarged endosomes. Although the function(s) of Alsin are not yet fully elucidated, experimental evidence supports a role for Alsin as a guanine exchange factor (GEF) on small guanosine triphosphatases (GTPases). GEFs are known to activate GTPases by stimulating GDP dissociation and GTP binding, thereby activating the GTPases, which act as switches in numerous signaling cascades. Previous studies have demonstrated that Alsin preferentially targets the Rab5 GTPase family, which is involved in signal transduction, trafficking, and vesicle formation, suggesting that Alsin may play a key role in endocytosis and cytoskeletal reorganization in mammalian neuronal cells [2][3][4]. Alterations in the Alsin protein, ranging from point mutations to complete deletion of structured domains, lead to diverse clinical scenarios in terms of onset, symptoms, and fatality. Therefore, the description of Alsin’s structure and its involvement in protein cascades related to cellular functions through protein–protein interactions is an essential topic that must be understood in depth for a rational view of the mechanisms behind the development of the above mentioned MNDs.
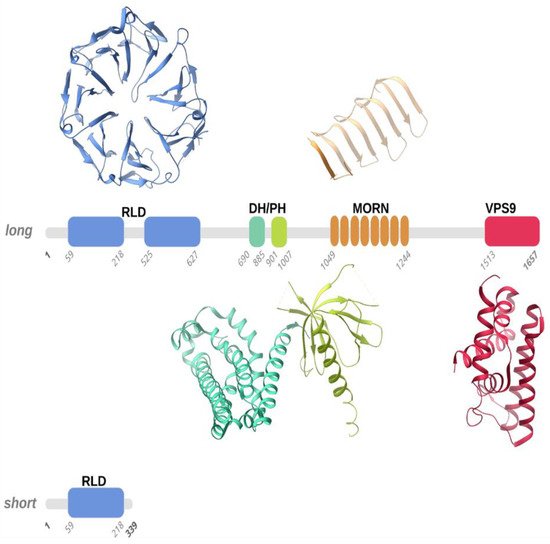
Figure 1. Schematic representation of the long and short forms of human Alsin. According to homology modelling, four structured domains, numbered according to the Uniprot Q96Q42 entry, were predicted in human Alsin, namely the RLD (RCC1-like domain), DH-PH (Dbl homology-pleckstrin homology), MORN (membrane occupation and recognition nexus), and VPS9 (vacuolar protein-sorting 9) domains. We used representative structures of these domains (PDB entries 1A12, 2Z0Q, 6T4D, and 2OT3) to illustrate the 3D folds that the RLD, DH-PH, MORN, and VPS9 domains predicted in human Alsin, respectively, may adopt. Structured domains are shown as ribbons with the same color code as the one utilized for the sequence schematic representation.
1.1. Alsin Molecular Structure
Alsin is a protein constituted by three putative GEF domains [5][2][6]: the RCC1-like domain (RLD) at the N-terminus, the central B cell lymphoma (Dbl) homology (DH) and pleckstrin-homology (PH) domain, and the C-terminal vacuolar protein-sorting 9 (VPS9) domain. Additionally, eight consecutive membrane occupation and recognition nexus (MORN) motifs are inserted in between the DH/PH and VPS9 domains [2][7] (Figure 1).
Interestingly, the RLD, DH/PH, and VPS9 domains have shown GEF activity, supporting the role of Alsin as a regulator and activator of multiple small GTPases [4][8]. Moreover, Alsin’s structured domains seem to drive the subcellular localization of protein. More specifically, studies on truncated variants revealed that C-terminal-truncated variants are mainly cytoplasmic, while N-terminal-truncated variants display endosomal localization [9][10].
1.1.1. RLD Domain and the Intrinsically Disordered Domain
Alsin’s N-terminal RCC1-like domain (RLD) shares homology with the RCC1 protein (Regulator of Chromosome Condensation 1-protein) [11][12]. Although clear experimental evidence is still lacking, homology modeling suggested that the Alsin RLD domain builds up a seven-bladed propeller by means of RCC1-like sequences of ~50-60 folded residues (propeller blades), which are stabilized by inter-blade disulphide bridges (Figure 1). Each blade of the propeller is composed of 4 β-strands connected in tandem by long loops: the N-terminal strand faces the central propeller tunnel, the second strand from one blade interacts with the third strand from the adjacent blade, and the fourth and shorter strand lies on the propeller surface. A conserved VyxWGT consensus sequence is observed on the third blade, which seems to participate in hydrophobic interactions between the blades, while the strong sequence variability of the last strand suggests that it could be involved in protein–protein interactions. It is worth noting that in the first blade, the first two beta-strands are constituted by the C-terminal part of the RLD domain, while the third and fourth strands are formed by the domain’s N-terminal sequence. This structural peculiarity was hypothesized to facilitate the completion of the propeller structure, favoring a correct folding of the domain owing to the first beta-strands from blade 1 acting as a velcro with the last two strands. Furthermore, in human Alsin, a ~300 amino acid sequence, predicted to be an intrinsically disordered region (IDR) and not evolutionarily conserved, segments the RCC1-like repeats in two regions and is inserted between the first two strands and the last two strands of blade 5. Disordered insertion in eucaryotic proteins is common and a structural transition may occur upon ligand/protein partner binding, as observed for many cellular signaling processes [13][14]. Literature studies suggested that the Alsin IDR may have a fundamental role in the proper self-oligomerization and in the intracellular distribution of the protein [15]. RCC1 is present in various proteins as a GEF for Ran (Ras-related nuclear), a small GTP-binding protein that is predominantly located in the nucleus and implicated in the nuclear import of proteins with nuclear localization signals and chromatin condensation through the regulation of microtubule assembly [16][17]. However, the cytosolic localization of Alsin, the absence of observed activity on Ran GTPase, and the non-conservation of the 25 residues of RCC1 protein responsible for Ran-binding make it rather unlikely for the Alsin RLD domain to exert an activity on Ran. Additionally, although a wide variety of proteins display RCC1 domains, only RCC1 itself has demonstrated Ran guanine nucleotide-exchange activity [9][18]. Even though this issue is still under debate, it seems that the Alsin RLD domain may drive subcellular localization and endosomal association through protein–protein interactions [10][19].
A short variant of Alsin that only contains part of the RLD domain exists (Figure 1); hypotheses were made about the role of this short variant in the variable severity of clinical phenotypes and the protective role of lower motor neurons (LMN) from degeneration, but this latter speculation was not confirmed since although a patient carried both Alsin variants, he did not show signs of LMN involvement [10][11][19]. Further studies demonstrated that the predicted protein derived from the short form of ALS2, together with the disease-associated mutant forms, are intrinsically unstable and may, therefore, undergo proteasome-mediated degradation in cultured human cells, including cells sampled from a patient with ALS2 mutations [10].
1.1.2. DH and PH Domains
Another two structured regions, which are almost invariably present together in homologues proteins, are the Dbl homology (DH) domain and the pleckstrin-homology (PH) domain. In general, DH domains form an eleven-α-helix bundle of ~200 residues, while PH domains are usually ~100 residue-modular domains composed of two perpendicular anti-parallel β-sheets, followed by a C-terminal amphipathic helix (Figure 1) [20]. Notably, the identification of the PH domain may not be an easy task owing to the versatility in lengths and sequence for loops connecting the β-strands. It was demonstrated that PH domains can bind phosphatidylinositol within biological membranes and proteins, such as the β/γ subunits of heterotrimeric G proteins [21], which belong to a family of proteins involved in signal transduction and amplification and act as molecular switches through their interaction with G-protein-coupled receptors (GPCRs). Another example of a protein-embedding phosphatidylinositol that can be bound by PH domains is the protein kinase C [22] that regulates numerous cellular responses such as gene expression, protein secretion, cell proliferation, and inflammatory response. These interactions allow PH domains to participate in the recruitment of proteins to different membranes to address them to appropriate cellular compartments or enable them to interact with other components of the signal transduction pathways. The fact that PH domains invariably exists in the presence of DH domains makes it reasonable to think that a functional interdependence exists between these domains. Moreover, it was demonstrated that DH/PH domains accommodate within the DH/PH domain junction interface [23][24] and, notably, that Rho proteins are involved in the organization of the actin cytoskeleton, in neuronal morphogenesis, and in various signaling cascades [8]. Previous in vivo and in vitro studies suggested that Alsin DH/PH domains catalyze GEF on both Rab5 and Rac1 [17], but although it was demonstrated that DH/PH domains directly and specifically interact with Rac1 [25], further tests on COS-7 and Hela cells showed that Rac1 GEF activity in these cells is not relevant [25].
1.1.3. MORN and VPS9 Domain
The VPS9 domain is common to numerous mammalian proteins and, more precisely, to GDP-GTP, which is involved in the activation of Rab5, a protein belonging to the Rab protein family of signal-transducing GTPases that cycle between active GTP-bound and inactive GDP-bound forms. This domain is composed of about 140 residues and catalyzes nucleotide exchange on the Rab5 GTPase, a regulator of endocytosis and endosome formation [26]. From a structural point of view, the VPS9 domain forms a layered fold of six α-helices (Figure 1). Notably, in the Rabex5 VPS9 domain, which shares homology with the Alsin VPS9 domain, the second and third helices form a helical hairpin that supports the four other helices [27]. Mutations of conserved residues (Asp 313, Pro 317, Tyr354, and Tyr 357) from the fourth and sixth helices hampered GEF activity on Rab5 and Rab21, suggesting that these two helices and the loop at the N-terminus of the fourth helix are the preferential interaction sites for Rab5 and Rab21 GTPases on the VPS9 domain [27].
The MORN motif is a tandem array of eight membrane occupation and recognition nexus motifs of 23 amino acids each [8] found in repeats in several proteins (Figure 1). Its function is unknown, but the repeats suggest involvement in Rab5-GEF activity through an association of Alsin protein with intracellular membranes [3]. Alsin MORN motifs and the VPS9 domain seem to work in synergy to drive endosome enlargement: in fact, a study on truncated variants showed that the lack of either the MORN motifs or the VPS9 domain abolished the GEF activity on Rab5 in vitro, whereas RLD-truncated variants promoted the formation of enlarged endosomes [9]. Moreover, the MORN/VPS9 domains are involved in Alsin self-interactions. Experimental evidence, using truncated variants in yeast cells, showed that the 1233-1351 and 1351-1454 regions are indispensable for Alsin homo-oligomerization. Additionally, it showed that although Rab5 binding to MORN/VPS9 is maintained if Alsin homo-association is hampered, the GEF activity on Rab5 is abolished [12].
The C-terminal half, including the MORN/VPS9 domains, is also involved in the endosomal localization of ALS2, while RLD in the long form of Alsin, after Rac1 binding, has an inhibitory role preventing ALS2 from being re-sequestrated by the self-interaction with RLD and MORN/VPS9 domains due to interaction with membrane ruffles [25]. It was found that the N-terminus with truncated ALS2, which therefore only contained the MORN/VPS9 region, was still capable of homo-dimerizing or homo-oligomerizing in mammalian cells, demonstrating that the C-terminal region is essential for Alsin homo-association [25].
1.2. Alsin Mutations Lead to Signalling Pathways Alteration
As clarified in the previous sections, ALS2 mutations reverberate in changes in the sequence of the Alsin protein. Such sequence modifications could impact the structural organization of the protein with consequential influence on intra-protein inter-domain interactions or protein–protein interactions of either Alsin with itself or Alsin with other protein partners. Changes in molecular interaction modes can result in changes in binding affinity or kinetics. Such changes can drastically alter protein cascades, strongly impacting cellular function. Although there are still not enough studies at the molecular level that can provide insight into the mechanisms of correlation between structure and function in Alsin, there are several experimental studies that have been able to assess Alsin’s misfunctions in the presence of specific molecular alterations, e.g., mutations. These studies are rationalized in the following sections.
1.2.1. Alsin’s RLD Domain and Altered Trafficking of AMPA Receptors
One of the causes of the degeneration of motor neurons involved in IAHSP is glutamate-mediated excitotoxicity, a pathological process that leads to neuron damage and death upon over-activation of receptors; more precisely, the α-amino-3-hydroxy-5-methylisoxazole-4-propio-nate (AMPA) glutamate receptors. AMPA glutamate receptors are ligand-dependent ion channels, allowing ions such as N+, K+, Cl-, and Ca2+ to pass through the neuron membrane upon binding of a neurotransmitter [28]. AMPA glutamate receptors are homo- or hetero-tetramers consisting of a combination of GluR1, GluR2, GluR3, and GluR4 subunits. Notably, both GluR1 and GluR2 play an important role in synaptic plasticity. Moreover, neurons that are exempt from GluR2-containing receptors are more exposed to excitotoxicity. The GruR2 subunit is usually located on the synaptic surface owing to its interaction with the Glutamate receptor-interacting protein 1 (GRIP1), responsible for GluR2 transport to dendrites in both postsynaptic membrane and intracellular compartments, to form the AMPA-glutamate receptor [19][29][30]. GRIP1 interacts with Alsin’s N-terminal RLD domain both in vitro and in vivo [19][29]. This is true not only for the long form of Alsin but also for the short form, which contains a partial RLD domain and also interacts with GRIP1, although in a weaker fashion [29]. A decrease in GluR2 and altered distribution of GRIP1 is observed upon the loss of Alsin. [19][29][30]. The experimental data, therefore, suggest that Alsin might modulate the traffic of AMPA receptors thanks to its interaction with GRIP1, and also that it may play a neuroprotective role against excitotoxicity and, therefore, this pathway may be envisioned as a potential therapeutic target for the treatment of Alsin and motor neuron pathologies [8][19][29][30].
1.2.2. Alsin Neuroprotective Role against SOD1 Mutations
It is firmly established that the mutations on chromosome 21q22.1 encoding for the Cu/Zn-superoxide dismutase (SOD1) can induce the degeneration of motor neurons, since it has been shown that mutated SOD1 acquires toxic properties [8][31][32][33]. SOD1 is a very abundant protein within the central nervous system; it constitutes 1% of all brain proteins [32][34]. Although the effect of SOD1 mutation is not yet fully understood, it is known that it leads to oxidative stress, mitochondrial dysfunction, excitotoxicity, protein aggregation, and inflammation. These alterations are not mutually exclusive and they may contribute to motor neuron degeneration [32]. Overexpression of Alsin may play a neuroprotective role in motor neurons by inhibiting the toxicity induced by SOD1 mutations through direct interaction between Alsin and mutated SOD1 [8]. Interestingly the DH/PH domain plays a dominant role in the above-mentioned interaction. It is worth noting that the above-highlighted Alsin–SOD1 interaction does not occur in the presence of wild-type SOD1 [8][32][33][35][36][37]. Moreover, ASL2 and SOD1 may share some functional characteristics. This is suggested by recent studies on genetically modified mice (ALS2-mutated mice and SOD1-mutated mice) showing altered toxicity due to inhibitors of mitochondrial complex 1 [31][35]. In a greater detail, transgenic mice endowed with Alsin-deprived neurons have been shown to be more sensitive to oxidative stress and to cell death induced by paraquat, an herbicide that is extremely toxic for cells. Taken together, the findings suggest that although the absence of Alsin may not be the sole factor inducing neurodegeneration in mice, it causes enhanced sensitivity to oxidative stress [19][36]. A clear neuroprotective role of Alsin against the degeneration of motor neurons induced by SOD1 mutations has not yet been demonstrated in vivo [31][36]. Therefore, a neuroprotective role has been established for Alsin in the presence of mutated SOD1 but not in the case of other neurotoxic insults [33][37]. Further evidence in favor of Alsin’s neuroprotective role is the observation that mutated ALS2 and SOD1 are co-localized, which was made by means of immunohistochemical analyses on transgenic rodents [33].
1.2.3. Alsin DH/PH Stimulates Rac1-PAK Binding and Neuron Growth
Interestingly, Alsin colocalizes with Filamentous actin (F-actin) inside the ruffles of the growth cone membrane and on F-actin-coated cone vesicles [38][25][39][40]. A recent study highlighted how Alsin’s DH/PH domain is essential for the activation of signaling pathways related to (i) GTPases such as Rho, and Rac1, and (ii) for p21-activated kinase (PAK1) [39]. The above-mentioned signaling pathways are known to promote neuron growth and development. Recent studies suggested that Alsin overexpression does not affect the development of the axons and dendrites of neurons. Moreover, experiments also showed how the length of neurites in wild-type mice is significantly increased, while in mice endowed with DH domain-mutated Alsin, neurite growth is not altered [25][39].
1.2.4. Alsin VPS9: Endosomal Trafficking and Rab5-Mediated Mechanisms
Endosomal trafficking is a recurrently affected pathway in several neurodegenerative diseases. In the following, we describe a number of sources of evidence that point towards a role of Alsin in the process involving Rab5 and suggest that the absence of Alsin or its mutations affect endosomal trafficking at different levels. Neurons use endocytosis to communicate with each other or with other tissues and to survive [30]. Several in vitro studies support the fact that Alsin dysfunction affects endosomal trafficking owing to the alteration of the VPS9 domain Rab5-mediated pathway [1][38][9][10][30][40][41]. Rab5 plays a fundamental role in the regulation of organelle binding, endosomal fusion, and motility in endocytosis. To prove this, experiments were performed where whole Alsin and truncated forms were found to be expressed [19]. It was observed that all forms were capable of triggering the release of GDP from Rab5, leading to endosomal budding [19]. Furthermore, overexpression of full-length Alsin results in a less efficient stimulation of endosomal fusion by Rab5 compared to RLD-domain-truncated Alsin, suggesting that the RLD domain can prevent Alsin from associating with early endosomes, thus acting as a negative regulator of Rab5-mediated endosomal fusion [38][9][19][30]. Interestingly, in ALS2 knockout homozygous (ALS2(−;−)) mouse neurons, an excessive amount of positive Rab5 vesicles and enlarged endosomes were observed, accompanied by a substantial decrease in endosomal motility [19][30]. Moreover, ALS2(−;−) mice neurons displayed reduced cell bodies and axons compared to those of wild-type ones; similar effects were observed regarding cerebellar weight and total brain weight [40]. This evidence would support the idea that malfunctioning or absent Alsin may cause a blockage or slowing of the endosomal trafficking of neurotrophic receptors and lead to decreased neurotrophic support. Even the primary neurons’ function was shown to be altered due to Alsin dysfunction since ALS2(−;−) mice suffer from disturbance of the endosomal transport of insulin-like growth factor (IGF1) and brain-derived neurotrophic factor receptors [9][19][30][41]. Moreover, Alsin is gathered inside the membranous compartments during micropinocytosis thanks to the activation of Rac1 [38][25][40], supporting the role of Alsin in mediating the fusion between early endosomes and macropinosomes. More specifically, the RLD domain drives the localization of Alsin and, therefore, it is essential for the recycling of Alsin in membranes [9][10][40]. Furthermore, missense mutants, despite displaying Rab5-GEF activity in vitro, are unable to localize Alsin correctly within early endosomes and/or macropinosomes even though the Rac1-induced micropinocytosis are active [25]. All the missense mutants passed from the cytosol to membrane ruffles, but they did not reach the endosomes or macropinosomes [38]. This observation suggests that the homo-tetrameric structure of Alsin may be required to activate this pathway; this means that a disordered Alsin structure leads to the loss of normal cellular functions [38]. Further support for the involvement of Alsin in endolysosomal trafficking is given by experimental evidence of Alsin colocalization with p62/LC3-positive autophagosomes [42].
In light of this, it has been suggested that Alsin may play a role in the maturation of autophagosomes into late endosomes and eventually participate in lysosome formation by accompanying Rab7 recruitment.
In conclusion, it is not yet possible to establish a comprehensive and consistent picture of the specific activity of Alsin in endosomal trafficking, but the data reported above unequivocally show that the absence of Alsin or certain mutations drive toward a plethora of effects in this process.
References
- Hadano, S.; Benn, S.C.; Kakuta, S.; Otomo, A.; Sudo, K.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Shefner, J.M.; Cox, G.A.; et al. Mice deficient in the Rab5 guanine nucleotide exchange factor ALS2/alsin exhibit age-dependent neurological deficits and altered endosome trafficking. Hum. Mol. Genet. 2006, 15, 233–250.
- Helal, M.; Mazaheri, N.; Shalbafan, B.; Malamiri, R.A.; Dilaver, N.; Buchert, R.; Mohammadiasl, J.; Golchin, N.; Sedaghat, A.; Mehrjardi, M.Y.V.; et al. Clinical presentation and natural history of infantile-onset ascending spastic paralysis from three families with an ALS2 founder variant. Neurol. Sci. 2018, 39, 1917–1925.
- Wakil, S.M.; Ramzan, K.; Abuthuraya, R.; Hagos, S.; Al-Dossari, H.; Al-Omar, R.; Murad, H.; Chedrawi, A.; Al-Hassnan, Z.N.; Finsterer, J.; et al. Infantile-onset ascending hereditary spastic paraplegia with bulbar involvement due to the novel ALS2 mutation c.2761C > T. Gene 2014, 536, 217–220.
- Sztriha, L.; Panzeri, C.; Kálmánchey, R.; Szabó, N.; Endreffy, E.; Túri, S.; Baschirotto, C.; Bresolin, N.; Vekerdy, Z.; Bassi, M. First case of compound heterozygosity in ALS2 gene in infantile-onset ascending spastic paralysis with bulbar involvement. Clin. Genet. 2008, 73, 591–593.
- Verschuuren-Bemelmans, C.C.; Winter, P.; Sival, D.A.; Elting, J.-W.; Brouwer, O.F.; Müller, U. Novel homozygous ALS2 nonsense mutation (p.Gln715X) in sibs with infantile-onset ascending spastic paralysis: The first cases from northwestern Europe. Eur. J. Hum. Genet. 2008, 16, 1407–1411.
- Kress, J.A.; Kühnlein, P.; Winter, P.; Ludolph, A.C.; Kassubek, J.; Müller, U.; Sperfeld, A.-D. Novel mutation in theALS2 gene in juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2005, 58, 800–803.
- Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Ikeda, J.-E.; Hadano, S. Defective relocalization of ALS2/alsin missense mutants to Rac1-induced macropinosomes accounts for loss of their cellular function and leads to disturbed amphisome formation. FEBS Lett. 2011, 585, 730–736.
- Hadano, S.; Kunita, R.; Otomo, A.; Suzuki-Utsunomiya, K.; Ikeda, J.-E. Molecular and cellular function of ALS2/alsin: Implication of membrane dynamics in neuronal development and degeneration. Neurochem. Int. 2007, 51, 74–84.
- Otomo, A. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003, 12, 1671–1687.
- Yamanaka, K.; Vande Velde, C.; Eymard-Pierre, E.; Bertini, E.; Boespflug-Tanguy, O.; Cleveland, D.W. Unstable mutants in the peripheral endosomal membrane component ALS2 cause early-onset motor neuron disease. Proc. Natl. Acad. Sci. USA 2003, 100, 16041–16046.
- Millecamps, S.; Gentil, B.J.; Gros-Louis, F.; Rouleau, G.; Julien, J.P. Alsin is partially associated with centrosome in human cells. Biochim. Biophys. Acta Mol. Cell Res. 2005, 1745, 84–100.
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki, K.; Showguchi-Miyata, J.; Yanagisawa, Y.; Hadano, S.; Ikeda, J.-E. Homo-oligomerization of ALS2 through Its Unique Carboxyl-terminal Regions Is Essential for the ALS2-associated Rab5 Guanine Nucleotide Exchange Activity and Its Regulatory Function on Endosome Trafficking. J. Biol. Chem. 2004, 279, 38626–38635.
- Dasso, M. RCC1 in the cell cycle: The regulator of chromosome condensation takes on new roles. Trends Biochem. Sci. 1993, 18, 96–101.
- Soares, D.C.; Barlow, P.N.; Porteous, D.J.; Devon, R.S. An interrupted beta-propeller and protein disorder: Structural bioinformatics insights into the N-terminus of alsin. J. Mol. Model. 2009, 15, 113–122.
- Shimakura, K.; Sato, K.; Mitsui, S.; Ono, S.; Otomo, A.; Hadano, S. The N-terminal intrinsically disordered region mediates intracellular localization and self-oligomerization of ALS2. Biochem. Biophys. Res. Commun. 2021, 569, 106–111.
- Dasso, M. Running on Ran: Nuclear transport and the mitotic spindle. Cell 2001, 104, 321–324.
- Topp, J.D.; Gray, N.W.; Gerard, R.D.; Horazdovsky, B.F. Alsin is a Rab5 and Rac1 guanine nucleotide exchange factor. J. Biol. Chem. 2004, 279, 24612–24623.
- Hadjebi, O.; Casas-Terradellas, E.; Garcia-Gonzalo, F.R.; Rosa, J.L. The RCC1 superfamily: From genes, to function, to disease. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1467–1479.
- Cai, H.; Shim, H.; Lai, C.; Xie, C.; Lin, X.; Yang, W.J.; Chandran, J. ALS2/Alsin Knockout Mice and Motor Neuron Diseases. Neurodegener. Dis. 2008, 5, 359–366.
- Worthylake, D.K.; Rossman, K.L.; Sondek, J. Crystal Structure of the DH/PH Fragment of Dbs without Bound GTPase. Structure 2004, 12, 1079–1086.
- Wang, D.S.; Shaw, R.; Winkelmann, J.C.; Shaw, G. Binding of PH domains of β-adrenergic receptor kinase and β-spectrin to WD40/β-transducin repeat containing regions of the β-subunit of trimeric G-proteins. Biochem. Biophys. Res. Commun. 1994, 203, 29–35.
- Yao, L.; Kawakami, Y.; Kawakami, T. The pleckstrin homology domain of Bruton tyrosine kinase interacts with protein kinase C. Proc. Natl. Acad. Sci. USA 1994, 91, 9175–9179.
- Aghazadeh, B.; Zhu, K.; Kubiseski, T.J.; Liu, G.A.; Pawson, T.; Zheng, Y.; Rosen, M.K. Structure and mutagenesis of the Dbl homology domain. Nat. Struct. Biol. 1998, 5, 1098–1107.
- Soisson, S.M.; Nimnual, A.S.; Uy, M.; Bar-Sagi, D.; Kuriyan, J. Crystal structure of the Dbl and pleckstrin homology domains from the human Son of sevenless protein. Cell 1998, 95, 259–268.
- Kunita, R.; Otomo, A.; Mizumura, H.; Suzuki-Utsunomiya, K.; Hadano, S.; Ikeda, J.-E. The Rab5 Activator ALS2/alsin Acts as a Novel Rac1 Effector through Rac1-activated Endocytosis. J. Biol. Chem. 2007, 282, 16599–16611.
- Carney, D.S.; Davies, B.A.; Horazdovsky, B.F. Vps9 domain-containing proteins: Activators of Rab5 GTPases from yeast to neurons. Trends Cell Biol. 2006, 16, 27–35.
- Delprato, A.; Merithew, E.; Lambright, D.G. Structure, exchange determinants, and family-wide Rab specificity of the tandem helical bundle and Vps9 domains of Rabex-5. Cell 2004, 118, 607–617.
- Kwak, S.; Weiss, J.H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 2006, 16, 281–287.
- Lai, C.; Xie, C.; McCormack, S.G.; Chiang, H.-C.; Michalak, M.K.; Lin, X.; Chandran, J.; Shim, H.; Shimoji, M.; Cookson, M.R.; et al. Amyotrophic Lateral Sclerosis 2-Deficiency Leads to Neuronal Degeneration in Amyotrophic Lateral Sclerosis through Altered AMPA Receptor Trafficking. J. Neurosci. 2006, 26, 11798–11806.
- Lai, C.; Xie, C.; Shim, H.; Chandran, J.; Howell, B.W.; Cai, H. Regulation of endosomal motility and degradation by amyotrophic lateral sclerosis 2/alsin. Mol. Brain 2009, 2, 23.
- Lin, X.; Shim, H.; Cai, H. Deficiency in the ALS2 gene does not affect the motor neuron degeneration in SOD1G93A transgenic mice. Neurobiol. Aging 2007, 28, 1628–1630.
- Shaw, P.J. Molecular and cellular pathways of neurodegeneration in motor neurone disease. J. Neurol. Neurosurg. Psychiatry 2005, 76, 1046–1057.
- Kanekura, K.; Hashimoto, Y.; Niikura, T.; Aiso, S.; Matsuoka, M.; Nishimoto, I. Alsin, the Product of ALS2 Gene, Suppresses SOD1 Mutant Neurotoxicity through RhoGEF Domain by Interacting with SOD1 Mutants. J. Biol. Chem. 2004, 279, 19247–19256.
- Rakhit, R.; Chakrabartty, A. Structure, folding, and misfolding of Cu, Zn superoxide dismutase in amyotrophic lateral sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1025–1037.
- Chandran, J.; Ding, J.; Cai, H. Alsin and the molecular pathways of amyotrophic lateral sclerosis. Mol. Neurobiol. 2007, 36, 224–231.
- Cai, H. Loss of ALS2 Function Is Insufficient to Trigger Motor Neuron Degeneration in Knock-Out Mice but Predisposes Neurons to Oxidative Stress. J. Neurosci. 2005, 25, 7567–7574.
- Kanekura, K.; Hashimoto, Y.; Kita, Y.; Sasabe, J.; Aiso, S.; Nishimoto, I.; Matsuoka, M. A Rac1/Phosphatidylinositol 3-Kinase/Akt3 Anti-apoptotic Pathway, Triggered by AlsinLF, the Product of the ALS2 Gene, Antagonizes Cu/Zn-superoxide Dismutase (SOD1) Mutant-induced Motoneuronal Cell Death. J. Biol. Chem. 2005, 280, 4532–4543.
- Sato, K.; Otomo, A.; Ueda, M.T.; Hiratsuka, Y.; Suzuki-Utsunomiya, K.; Sugiyama, J.; Murakoshi, S.; Mitsui, S.; Ono, S.; Nakagawa, S.; et al. Altered oligomeric states in pathogenic ALS2 variants associated with juvenile motor neuron diseases cause loss of ALS2-mediated endosomal function. J. Biol. Chem. 2018, 293, 17135–17153.
- Tudor, E.L.; Perkinton, M.S.; Schmidt, A.; Ackerley, S.; Brownlees, J.; Jacobsen, N.J.O.; Byers, H.L.; Ward, M.; Hall, A.; Leigh, P.N.; et al. ALS2/Alsin Regulates Rac-PAK Signaling and Neurite Outgrowth. J. Biol. Chem. 2005, 280, 34735–34740.
- Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Mizumura, H.; Onoe, K.; Osuga, H.; Hadano, S.; Ikeda, J.-E. ALS2/alsin deficiency in neurons leads to mild defects in macropinocytosis and axonal growth. Biochem. Biophys. Res. Commun. 2008, 370, 87–92.
- Devon, R.S.; Orban, P.C.; Gerrow, K.; Barbieri, M.A.; Schwab, C.; Cao, L.P.; Helm, J.R.; Bissada, M.; Cruz-Aguado, R.; Davidson, T.L.; et al. Als2-deficient mice exhibit disturbances in endosome trafficking associated with motor behavioral abnormalities. Proc. Natl. Acad. Sci. USA 2006, 103, 9595–9600.
- Hadano, S.; Otomo, A.; Kunita, R.; Suzuki-Utsunomiya, K.; Akatsuka, A.; Koike, M.; Aoki, M.; Uchiyama, Y.; Itoyama, Y.; Ikeda, J.-E. Loss of ALS2/Alsin Exacerbates Motor Dysfunction in a SOD1H46R-Expressing Mouse ALS Model by Disturbing Endolysosomal Trafficking. PLoS ONE 2010, 5, e9805.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
824
Revisions:
2 times
(View History)
Update Date:
20 Jan 2022
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No