 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Seonghwan Hwang | + 5144 word(s) | 5144 | 2022-01-18 06:40:22 | | | |
| 2 | Bruce Ren | Meta information modification | 5144 | 2022-01-20 02:05:01 | | |
Video Upload Options
Alcoholic liver disease (ALD) is characterized by the injury, inflammation, and scarring in the liver owing to excessive alcohol consumption. Currently, ALD is a leading cause for liver transplantation. Therefore, extensive studies (in vitro, in experimental ALD models and in humans) are needed to elucidate pathological features and pathogenic mechanisms underlying ALD. Notably, oxidative changes in the liver have been recognized as a signature trait of ALD. Progression of ALD is linked to the generation of highly reactive free radicals by reactions involving ethanol and its metabolites. Furthermore, hepatic oxidative stress promotes tissue injury and, in turn, stimulates inflammatory responses in the liver, forming a pathological loop that promotes the progression of ALD. Accordingly, accumulating further knowledge on the relationship between oxidative stress and inflammation may help establish a viable therapeutic approach for treating ALD.
1. Introduction
2. Oxidative Stress-Related Pathogenic Mechanisms of ALD
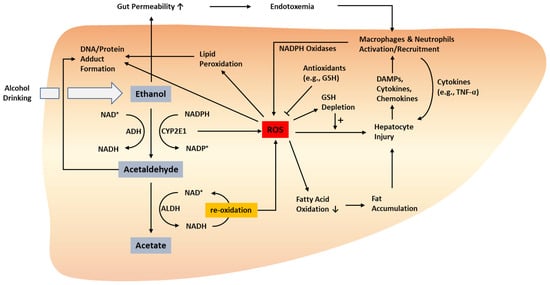
2.1. Alcohol-Induced Hepatocyte Injury
2.2. Immune Cells Mediating the Crosstalk between Oxidative Stress and Inflammation in ALD
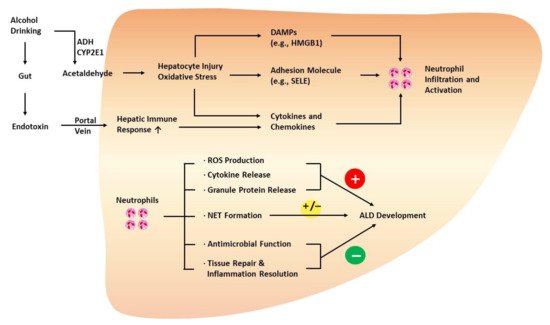
2.2.1. Neutrophils
2.2.2. Macrophages
2.2.3. Other Types of Immune Cells
2.3. The Role of MicroRNAs in the Crosstalk between Oxidative Stress and Inflammation in ALD
| microRNA | Status in ALD | Targets | Effects | References |
|---|---|---|---|---|
| Let-7b | Up | TLR7 activation | ↑hepatic inflammatory response | [201] |
| miR-150-5p | Up | CISH | ↑FADD-mediated programmed cell death | [202] |
| miR-155 | Up | Cebpb | ↑M1 macrophage polarization ↑fatty liver |
[203][204][205][206] |
| miR-181b | Up | PIAS1 | oxidative stress and inflammation | [197][207] |
| miR-182 | Up | SLC1A1 CFL1 |
↑liver injury and inflammation | [197] |
| miR-214 | Up | GSR POR |
↑oxidative stress | [208] |
| miR-223 | Up | IL-6 | ↓oxidative stress | [117] |
| miR-540 | Up | PPARα, PMP70, ACOX1, CPT1a | ↑hepatic steatosis | [209] |
| miR-148a | Down | TXNIP | ↑TXNIP-dependent inflammasome activation ↑ADH4 and CYP2B6 |
[10][210][211] |
| miR-219a-5p | Down | P66shc | ↑oxidative stress | [212] |
References
- Piano, M.R. Alcohol’s Effects on the Cardiovascular System. Alcohol Res. 2017, 38, 219–241.
- Obad, A.; Peeran, A.; Little, J.I.; Haddad, G.E.; Tarzami, S.T. Alcohol-Mediated Organ Damages: Heart and Brain. Front. Pharmacol. 2018, 9, 81.
- Pasala, S.; Barr, T.; Messaoudi, I. Impact of Alcohol Abuse on the Adaptive Immune System. Alcohol Res. 2015, 37, 185–197.
- Barr, T.; Helms, C.; Grant, K.; Messaoudi, I. Opposing effects of alcohol on the immune system. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 65, 242–251.
- Szabo, G.; Saha, B. Alcohol’s Effect on Host Defense. Alcohol Res. 2015, 37, 159–170.
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585.
- Mandrekar, P.; Bataller, R.; Tsukamoto, H.; Gao, B. Alcoholic hepatitis: Translational approaches to develop targeted therapies. Hepatology 2016, 64, 1343–1355.
- Chacko, K.R.; Reinus, J. Spectrum of Alcoholic Liver Disease. Clin. Liver Dis. 2016, 20, 419–427.
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161.
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol dysregulates miR-148a in hepatocytes through FoxO1, facilitating pyroptosis via TXNIP overexpression. Gut 2019, 68, 708–720.
- Sakhuja, P. Pathology of alcoholic liver disease, can it be differentiated from nonalcoholic steatohepatitis? World J. Gastroenterol. 2014, 20, 16474–16479.
- Lackner, C.; Tiniakos, D. Fibrosis and alcohol-related liver disease. J. Hepatol. 2019, 70, 294–304.
- Chayanupatkul, M.; Liangpunsakul, S. Alcoholic hepatitis: A comprehensive review of pathogenesis and treatment. World J. Gastroenterol. 2014, 20, 6279–6286.
- Marroni, C.A.; Fleck, A.M., Jr.; Fernandes, S.A.; Galant, L.H.; Mucenic, M.; de Mattos Meine, M.H.; Mariante-Neto, G.; Brandão, A.B.M. Liver transplantation and alcoholic liver disease: History, controversies, and considerations. World J. Gastroenterol. 2018, 24, 2785–2805.
- Chuncharunee, L.; Yamashiki, N.; Thakkinstian, A.; Sobhonslidsuk, A. Alcohol relapse and its predictors after liver transplantation for alcoholic liver disease: A systematic review and meta-analysis. BMC Gastroenterol. 2019, 19, 150.
- Lucey, M.R.; Mathurin, P.; Morgan, T.R. Alcoholic hepatitis. N. Engl. J. Med. 2009, 360, 2758–2769.
- Amini, M.; Runyon, B.A. Alcoholic hepatitis 2010: A clinician’s guide to diagnosis and therapy. World J. Gastroenterol. 2010, 16, 4905–4912.
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021, 18, 73–91.
- Louvet, A.; Mathurin, P. Alcoholic liver disease: Mechanisms of injury and targeted treatment. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 231–242.
- Sun, J.; Fu, J.; Li, L.; Chen, C.; Wang, H.; Hou, Y.; Xu, Y.; Pi, J. Nrf2 in alcoholic liver disease. Toxicol. Appl. Pharmacol. 2018, 357, 62–69.
- Donohue, T.M., Jr. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978.
- Ji, C. Advances and New Concepts in Alcohol-Induced Organelle Stress, Unfolded Protein Responses and Organ Damage. Biomolecules 2015, 5, 1099–1121.
- Cao, S.; Liu, M.; Sehrawat, T.S.; Shah, V.H. Regulation and functional roles of chemokines in liver diseases. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 630–647.
- Fujii, H.; Kawada, N. Fibrogenesis in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 8048–8054.
- Mantena, S.K.; King, A.L.; Andringa, K.K.; Landar, A.; Darley-Usmar, V.; Bailey, S.M. Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J. Gastroenterol. 2007, 13, 4967–4973.
- Speisky, H.; Kera, Y.; Penttilä, K.E.; Israel, Y.; Lindros, K.O. Depletion of hepatic glutathione by ethanol occurs independently of ethanol metabolism. Alcohol. Clin. Exp. Res. 1988, 12, 224–228.
- Jewell, S.A.; Di Monte, D.; Gentile, A.; Guglielmi, A.; Altomare, E.; Albano, O. Decreased hepatic glutathione in chronic alcoholic patients. J. Hepatol. 1986, 3, 1–6.
- Wu, D.; Cederbaum, A.I. Oxidative stress and alcoholic liver disease. Semin. Liver Dis. 2009, 29, 141–154.
- Zhu, H.; Jia, Z.; Misra, H.; Li, Y.R. Oxidative stress and redox signaling mechanisms of alcoholic liver disease: Updated experimental and clinical evidence. J. Dig. Dis. 2012, 13, 133–142.
- Lu, Y.; Zhuge, J.; Wang, X.; Bai, J.; Cederbaum, A.I. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology 2008, 47, 1483–1494.
- Cederbaum, A.I. Alcohol metabolism. Clin. Liver Dis. 2012, 16, 667–685.
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227.
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347.
- Esfandiari, F.; Medici, V.; Wong, D.H.; Jose, S.; Dolatshahi, M.; Quinlivan, E.; Dayal, S.; Lentz, S.R.; Tsukamoto, H.; Zhang, Y.H.; et al. Epigenetic regulation of hepatic endoplasmic reticulum stress pathways in the ethanol-fed cystathionine beta synthase-deficient mouse. Hepatology 2010, 51, 932–941.
- Ji, C.; Deng, Q.; Kaplowitz, N. Role of TNF-alpha in ethanol-induced hyperhomocysteinemia and murine alcoholic liver injury. Hepatology 2004, 40, 442–451.
- Galli, A.; Pinaire, J.; Fischer, M.; Dorris, R.; Crabb, D.W. The transcriptional and DNA binding activity of peroxisome proliferator-activated receptor alpha is inhibited by ethanol metabolism. A novel mechanism for the development of ethanol-induced fatty liver. J. Biol. Chem. 2001, 276, 68–75.
- Wagner, M.; Zollner, G.; Trauner, M. Nuclear receptors in liver disease. Hepatology 2011, 53, 1023–1034.
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16.
- Lieber, C.S. Ethanol metabolism, cirrhosis and alcoholism. Clin. Chim. Acta 1997, 257, 59–84.
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13.
- Edenberg, H.J.; McClintick, J.N. Alcohol Dehydrogenases, Aldehyde Dehydrogenases, and Alcohol Use Disorders: A Critical Review. Alcohol. Clin. Exp. Res. 2018, 42, 2281–2297.
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines 2020, 8, 50.
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15.
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254.
- Mailloux, R.J. Teaching the fundamentals of electron transfer reactions in mitochondria and the production and detection of reactive oxygen species. Redox. Biol. 2015, 4, 381–398.
- Zhao, M.; Matter, K.; Laissue, J.A.; Zimmermann, A. Copper/zinc and manganese superoxide dismutases in alcoholic liver disease: Immunohistochemical quantitation. Histol. Histopathol. 1996, 11, 899–907.
- Choi, D.W.; Kim, S.Y.; Kim, S.K.; Kim, Y.C. Factors involved in hepatic glutathione depletion induced by acute ethanol administration. J. Toxicol. Environ. Health A 2000, 60, 459–469.
- Lu, Y.; Cederbaum, A.I. CYP2E1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738.
- Leung, T.M.; Nieto, N. CYP2E1 and oxidant stress in alcoholic and non-alcoholic fatty liver disease. J. Hepatol. 2013, 58, 395–398.
- Bai, J.; Cederbaum, A.I. Overexpression of CYP2E1 in mitochondria sensitizes HepG2 cells to the toxicity caused by depletion of glutathione. J. Biol. Chem. 2006, 281, 5128–5136.
- Gouillon, Z.; Lucas, D.; Li, J.; Hagbjork, A.L.; French, B.A.; Fu, P.; Fang, C.; Ingelman-Sundberg, M.; Donohue, T.M., Jr.; French, S.W. Inhibition of ethanol-induced liver disease in the intragastric feeding rat model by chlormethiazole. Proc. Soc. Exp. Biol. Med. 2000, 224, 302–308.
- Ye, Q.; Lian, F.; Chavez, P.R.; Chung, J.; Ling, W.; Qin, H.; Seitz, H.K.; Wang, X.D. Cytochrome P450 2E1 inhibition prevents hepatic carcinogenesis induced by diethylnitrosamine in alcohol-fed rats. Hepatobiliary Surg. Nutr. 2012, 1, 5–18.
- Diesinger, T.; Buko, V.; Lautwein, A.; Dvorsky, R.; Belonovskaya, E.; Lukivskaya, O.; Naruta, E.; Kirko, S.; Andreev, V.; Buckert, D.; et al. Drug targeting CYP2E1 for the treatment of early-stage alcoholic steatohepatitis. PLoS ONE 2020, 15, e0235990.
- Sun, Q.; Zhang, W.; Zhong, W.; Sun, X.; Zhou, Z. Pharmacological inhibition of NOX4 ameliorates alcohol-induced liver injury in mice through improving oxidative stress and mitochondrial function. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2912–2921.
- Yang, C.F.; Zhong, Y.J.; Ma, Z.; Li, L.; Shi, L.; Chen, L.; Li, C.; Wu, D.; Chen, Q.; Li, Y.W. NOX4/ROS mediate ethanolinduced apoptosis via MAPK signal pathway in L02 cells. Int. J. Mol. Med. 2018, 41, 2306–2316.
- Miyata, T.; Nagy, L.E. Programmed cell death in alcohol-associated liver disease. Clin. Mol. Hepatol. 2020, 26, 618–625.
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063.
- Sastre, J.; Serviddio, G.; Pereda, J.; Minana, J.B.; Arduini, A.; Vendemiale, G.; Poli, G.; Pallardo, F.V.; Vina, J. Mitochondrial function in liver disease. Front. Biosci. 2007, 12, 1200–1209.
- Shalbueva, N.; Mareninova, O.A.; Gerloff, A.; Yuan, J.; Waldron, R.T.; Pandol, S.J.; Gukovskaya, A.S. Effects of oxidative alcohol metabolism on the mitochondrial permeability transition pore and necrosis in a mouse model of alcoholic pancreatitis. Gastroenterology 2013, 144, 437–446.
- Zelickson, B.R.; Benavides, G.A.; Johnson, M.S.; Chacko, B.K.; Venkatraman, A.; Landar, A.; Betancourt, A.M.; Bailey, S.M.; Darley-Usmar, V.M. Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim. Biophys. Acta 2011, 1807, 1573–1582.
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556.
- Zou, H.; Yang, R.; Hao, J.; Wang, J.; Sun, C.; Fesik, S.W.; Wu, J.C.; Tomaselli, K.J.; Armstrong, R.C. Regulation of the Apaf-1/caspase-9 apoptosome by caspase-3 and XIAP. J. Biol. Chem. 2003, 278, 8091–8098.
- Bratton, S.B.; Salvesen, G.S. Regulation of the Apaf-1-caspase-9 apoptosome. J. Cell Sci. 2010, 123, 3209–3214.
- Pastorino, J.G.; Shulga, N.; Hoek, J.B. TNF-alpha-induced cell death in ethanol-exposed cells depends on p38 MAPK signaling but is independent of Bid and caspase-8. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G503–G516.
- Milic, S.; Mikolasevic, I.; Orlic, L.; Devcic, E.; Starcevic-Cizmarevic, N.; Stimac, D.; Kapovic, M.; Ristic, S. The Role of Iron and Iron Overload in Chronic Liver Disease. Med. Sci. Monit. 2016, 22, 2144–2151.
- Anderson, E.R.; Taylor, M.; Xue, X.; Martin, A.; Moons, D.S.; Omary, M.B.; Shah, Y.M. The hypoxia-inducible factor-C/EBPα axis controls ethanol-mediated hepcidin repression. Mol. Cell. Biol. 2012, 32, 4068–4077.
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093.
- Harrison-Findik, D.D.; Schafer, D.; Klein, E.; Timchenko, N.A.; Kulaksiz, H.; Clemens, D.; Fein, E.; Andriopoulos, B.; Pantopoulos, K.; Gollan, J. Alcohol metabolism-mediated oxidative stress down-regulates hepcidin transcription and leads to increased duodenal iron transporter expression. J. Biol. Chem. 2006, 281, 22974–22982.
- Ioannou, G.N.; Dominitz, J.A.; Weiss, N.S.; Heagerty, P.J.; Kowdley, K.V. The effect of alcohol consumption on the prevalence of iron overload, iron deficiency, and iron deficiency anemia. Gastroenterology 2004, 126, 1293–1301.
- Purohit, V.; Russo, D.; Salin, M. Role of iron in alcoholic liver disease: Introduction and summary of the symposium. Alcohol 2003, 30, 93–97.
- Kohgo, Y.; Ohtake, T.; Ikuta, K.; Suzuki, Y.; Hosoki, Y.; Saito, H.; Kato, J. Iron accumulation in alcoholic liver diseases. Alcohol. Clin. Exp. Res. 2005, 29, 189s–193s.
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell. Biol. 2021, 22, 266–282.
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88.
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619.
- Wen, Q.; Liu, J.; Kang, R.; Zhou, B.; Tang, D. The release and activity of HMGB1 in ferroptosis. Biochem. Biophys. Res. Commun. 2019, 510, 278–283.
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1893–1900.
- Su, L.J.; Zhang, J.H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843.
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 73, 394–408.
- Mao, L.; Zhao, T.; Song, Y.; Lin, L.; Fan, X.; Cui, B.; Feng, H.; Wang, X.; Yu, Q.; Zhang, J.; et al. The emerging role of ferroptosis in non-cancer liver diseases: Hype or increasing hope? Cell Death Dis. 2020, 11, 518.
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651.
- Zhou, Z.; Ye, T.J.; DeCaro, E.; Buehler, B.; Stahl, Z.; Bonavita, G.; Daniels, M.; You, M. Intestinal SIRT1 Deficiency Protects Mice from Ethanol-Induced Liver Injury by Mitigating Ferroptosis. Am. J. Pathol. 2020, 190, 82–92.
- Zhou, Z.; Ye, T.J.; Bonavita, G.; Daniels, M.; Kainrad, N.; Jogasuria, A.; You, M. Adipose-Specific Lipin-1 Overexpression Renders Hepatic Ferroptosis and Exacerbates Alcoholic Steatohepatitis in Mice. Hepatol. Commun. 2019, 3, 656–669.
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630.
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176.
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62.
- Mueller, S.; Peccerella, T.; Qin, H.; Glassen, K.; Waldherr, R.; Flechtenmacher, C.; Straub, B.K.; Millonig, G.; Stickel, F.; Bruckner, T.; et al. Carcinogenic Etheno DNA Adducts in Alcoholic Liver Disease: Correlation with Cytochrome P-4502E1 and Fibrosis. Alcohol. Clin. Exp. Res. 2018, 42, 252–259.
- Lu, L.; Jiang, J.; Zhan, M.; Zhang, H.; Wang, Q.T.; Sun, S.N.; Guo, X.K.; Yin, H.; Wei, Y.; Liu, J.O.; et al. Targeting Neoantigens in Hepatocellular Carcinoma for Immunotherapy: A Futile Strategy? Hepatology 2021, 73, 414–421.
- Ma, J.; Cao, H.; Rodrigues, R.M.; Xu, M.; Ren, T.; He, Y.; Hwang, S.; Feng, D.; Ren, R.; Yang, P.; et al. Chronic-plus-binge alcohol intake induces production of proinflammatory mtDNA-enriched extracellular vesicles and steatohepatitis via ASK1/p38MAPKα-dependent mechanisms. JCI Insight 2020, 5, e136496.
- Wang, G.; Fu, Y.; Li, J.; Li, Y.; Zhao, Q.; Hu, A.; Xu, C.; Shao, D.; Chen, W. Aqueous extract of Polygonatum sibiricum ameliorates ethanol-induced mice liver injury via regulation of the Nrf2/ARE pathway. J. Food. Biochem. 2021, 45, e13537.
- Jiang, W.; Zhu, H.; Xu, W.; Liu, C.; Hu, B.; Guo, Y.; Cheng, Y.; Qian, H. Echinacea purpurea polysaccharide prepared by fractional precipitation prevents alcoholic liver injury in mice by protecting the intestinal barrier and regulating liver-related pathways. Int. J. Biol. Macromol. 2021, 187, 143–156.
- Sun, J.; Hong, Z.; Shao, S.; Li, L.; Yang, B.; Hou, Y.; Wang, H.; Xu, Y.; Zhang, Q.; Pi, J.; et al. Liver-specific Nrf2 deficiency accelerates ethanol-induced lethality and hepatic injury in vivo. Toxicol. Appl. Pharmacol. 2021, 426, 115617.
- Zhao, X.; Gong, L.; Wang, C.; Liu, M.; Hu, N.; Dai, X.; Peng, C.; Li, Y. Quercetin mitigates ethanol-induced hepatic steatosis in zebrafish via P2X7R-mediated PI3K/ Keap1/Nrf2 signaling pathway. J. Ethnopharmacol. 2021, 268, 113569.
- Wang, X.; Chang, X.; Zhan, H.; Zhang, Q.; Li, C.; Gao, Q.; Yang, M.; Luo, Z.; Li, S.; Sun, Y. Curcumin and Baicalin ameliorate ethanol-induced liver oxidative damage via the Nrf2/HO-1 pathway. J. Food Biochem. 2020, 44, e13425.
- Sabitha, R.; Nishi, K.; Gunasekaran, V.P.; Agilan, B.; David, E.; Annamalai, G.; Vinothkumar, R.; Perumal, M.; Subbiah, L.; Ganeshan, M. p-Coumaric acid attenuates alcohol exposed hepatic injury through MAPKs, apoptosis and Nrf2 signaling in experimental models. Chem. Biol. Interact. 2020, 321, 109044.
- Ge, X.; Antoine, D.J.; Lu, Y.; Arriazu, E.; Leung, T.M.; Klepper, A.L.; Branch, A.D.; Fiel, M.I.; Nieto, N. High mobility group box-1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J. Biol. Chem. 2014, 289, 22672–22691.
- Cai, Y.; Xu, M.J.; Koritzinsky, E.H.; Zhou, Z.; Wang, W.; Cao, H.; Yuen, P.S.; Ross, R.A.; Star, R.A.; Liangpunsakul, S.; et al. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight 2017, 2, e92634.
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104.
- Hoyt, L.R.; Randall, M.J.; Ather, J.L.; DePuccio, D.P.; Landry, C.C.; Qian, X.; Janssen-Heininger, Y.M.; van der Vliet, A.; Dixon, A.E.; Amiel, E.; et al. Mitochondrial ROS induced by chronic ethanol exposure promote hyper-activation of the NLRP3 inflammasome. Redox Biol. 2017, 12, 883–896.
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159.
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328.
- Kany, S.; Janicova, A.; Relja, B. Innate Immunity and Alcohol. J. Clin. Med. 2019, 8, 1981.
- Kim, A.; Bellar, A.; McMullen, M.R.; Li, X.; Nagy, L.E. Functionally Diverse Inflammatory Responses in Peripheral and Liver Monocytes in Alcohol-Associated Hepatitis. Hepatol. Commun. 2020, 4, 1459–1476.
- Xu, M.J.; Zhou, Z.; Parker, R.; Gao, B. Targeting inflammation for the treatment of alcoholic liver disease. Pharmacol. Ther. 2017, 180, 77–89.
- Mottaran, E.; Stewart, S.F.; Rolla, R.; Vay, D.; Cipriani, V.; Moretti, M.; Vidali, M.; Sartori, M.; Rigamonti, C.; Day, C.P.; et al. Lipid peroxidation contributes to immune reactions associated with alcoholic liver disease. Free Radic. Biol. Med. 2002, 32, 38–45.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017, 7, 373.
- Tan, H.Y.; Wang, N.; Li, S.; Hong, M.; Wang, X.; Feng, Y. The Reactive Oxygen Species in Macrophage Polarization: Reflecting Its Dual Role in Progression and Treatment of Human Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 2795090.
- Fialkow, L.; Wang, Y.; Downey, G.P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 2007, 42, 153–164.
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366.
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 113.
- Luk, G.D.; Silverman, A.L.; Giardiello, F.M. Biochemical markers in patients with familial colonic neoplasia. Semin. Surg. Oncol. 1987, 3, 126–132.
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594.
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175.
- Liu, K.; Wang, F.S.; Xu, R. Neutrophils in liver diseases: Pathogenesis and therapeutic targets. Cell. Mol. Immunol. 2021, 18, 38–44.
- Mathews, S.; Feng, D.; Maricic, I.; Ju, C.; Kumar, V.; Gao, B. Invariant natural killer T cells contribute to chronic-plus-binge ethanol-mediated liver injury by promoting hepatic neutrophil infiltration. Cell. Mol. Immunol. 2016, 13, 206–216.
- Bertola, A.; Park, O.; Gao, B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: A critical role for E-selectin. Hepatology 2013, 58, 1814–1823.
- Hwang, S.; Ren, T.; Gao, B. Obesity and binge alcohol intake are deadly combination to induce steatohepatitis: A model of high-fat diet and binge ethanol intake. Clin. Mol. Hepatol. 2020, 26, 586–594.e1.
- Li, M.; He, Y.; Zhou, Z.; Ramirez, T.; Gao, Y.; Gao, Y.; Ross, R.A.; Cao, H.; Cai, Y.; Xu, M.; et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47(phox)-oxidative stress pathway in neutrophils. Gut 2017, 66, 705–715.
- Lazaro, R.; Wu, R.; Lee, S.; Zhu, N.L.; Chen, C.L.; French, S.W.; Xu, J.; Machida, K.; Tsukamoto, H. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology 2015, 61, 129–140.
- Bertola, A.; Mathews, S.; Ki, S.H.; Wang, H.; Gao, B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat. Protoc. 2013, 8, 627–637.
- Blüml, S.; Rosc, B.; Lorincz, A.; Seyerl, M.; Kirchberger, S.; Oskolkova, O.; Bochkov, V.N.; Majdic, O.; Ligeti, E.; Stöckl, J. The oxidation state of phospholipids controls the oxidative burst in neutrophil granulocytes. J. Immunol. 2008, 181, 4347–4353.
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as emerging therapeutic targets. Nat. Rev. Drug. Discov. 2020, 19, 253–275.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23.
- Khanam, A.; Trehanpati, N.; Riese, P.; Rastogi, A.; Guzman, C.A.; Sarin, S.K. Blockade of Neutrophil’s Chemokine Receptors CXCR1/2 Abrogate Liver Damage in Acute-on-Chronic Liver Failure. Front. Immunol. 2017, 8, 464.
- Hwang, S.; He, Y.; Xiang, X.; Seo, W.; Kim, S.J.; Ma, J.; Ren, T.; Park, S.H.; Zhou, Z.; Feng, D.; et al. Interleukin-22 Ameliorates Neutrophil-Driven Nonalcoholic Steatohepatitis Through Multiple Targets. Hepatology 2020, 72, 412–429.
- Roh, Y.S.; Zhang, B.; Loomba, R.; Seki, E. TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 309, G30–G41.
- Lemmers, A.; Moreno, C.; Gustot, T.; Maréchal, R.; Degré, D.; Demetter, P.; de Nadai, P.; Geerts, A.; Quertinmont, E.; Vercruysse, V.; et al. The interleukin-17 pathway is involved in human alcoholic liver disease. Hepatology 2009, 49, 646–657.
- Ma, H.Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 2020, 72, 946–959.
- Mookerjee, R.P.; Stadlbauer, V.; Lidder, S.; Wright, G.A.; Hodges, S.J.; Davies, N.A.; Jalan, R. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology 2007, 46, 831–840.
- Yang, W.; Tao, Y.; Wu, Y.; Zhao, X.; Ye, W.; Zhao, D.; Fu, L.; Tian, C.; Yang, J.; He, F.; et al. Neutrophils promote the development of reparative macrophages mediated by ROS to orchestrate liver repair. Nat. Commun. 2019, 10, 1076.
- Tecchio, C.; Micheletti, A.; Cassatella, M.A. Neutrophil-derived cytokines: Facts beyond expression. Front. Immunol. 2014, 5, 508.
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514.
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56.
- Karakucuk, I.; Dilly, S.A.; Maxwell, J.D. Portal tract macrophages are increased in alcoholic liver disease. Histopathology 1989, 14, 245–253.
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665.
- Koop, D.R.; Klopfenstein, B.; Iimuro, Y.; Thurman, R.G. Gadolinium chloride blocks alcohol-dependent liver toxicity in rats treated chronically with intragastric alcohol despite the induction of CYP2E1. Mol. Pharmacol. 1997, 51, 944–950.
- Rao, R. Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology 2009, 50, 638–644.
- Zhou, Z.; Zhong, W. Targeting the gut barrier for the treatment of alcoholic liver disease. Liver Res. 2017, 1, 197–207.
- Keshavarzian, A.; Farhadi, A.; Forsyth, C.B.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J. Hepatol. 2009, 50, 538–547.
- Singal, A.K.; Shah, V.H. Current trials and novel therapeutic targets for alcoholic hepatitis. J. Hepatol. 2019, 70, 305–313.
- Gottfried, E.B.; Korsten, M.A.; Lieber, C.S. Alcohol-induced gastric and duodenal lesions in man. Am. J. Gastroenterol. 1978, 70, 587–592.
- Bode, J.C.; Knüppel, H.; Schwerk, W.; Lorenz-Meyer, H.; Dürr, H.K. Quantitative histomorphometric study of the jejunal mucosa in chronic alcoholics. Digestion 1982, 23, 265–270.
- Tamai, H.; Kato, S.; Horie, Y.; Ohki, E.; Yokoyama, H.; Ishii, H. Effect of acute ethanol administration on the intestinal absorption of endotoxin in rats. Alcohol. Clin. Exp. Res. 2000, 24, 390–394.
- Lambert, J.C.; Zhou, Z.; Wang, L.; Song, Z.; McClain, C.J.; Kang, Y.J. Prevention of alterations in intestinal permeability is involved in zinc inhibition of acute ethanol-induced liver damage in mice. J. Pharmacol. Exp. Ther. 2003, 305, 880–886.
- Asai, K.; Buurman, W.A.; Reutelingsperger, C.P.; Schutte, B.; Kaminishi, M. Low concentrations of ethanol induce apoptosis in human intestinal cells. Scand. J. Gastroenterol. 2003, 38, 1154–1161.
- Amin, P.B.; Diebel, L.N.; Liberati, D.M. Dose-dependent effects of ethanol and E. coli on gut permeability and cytokine production. J. Surg. Res. 2009, 157, 187–192.
- Elamin, E.; Masclee, A.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Pieters, H.J.; Dekker, J.; Jonkers, D. Fatty acid ethyl esters induce intestinal epithelial barrier dysfunction via a reactive oxygen species-dependent mechanism in a three-dimensional cell culture model. PLoS ONE 2013, 8, e58561.
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008.
- Atkinson, K.J.; Rao, R.K. Role of protein tyrosine phosphorylation in acetaldehyde-induced disruption of epithelial tight junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1280–G1288.
- Rao, R.K.; Baker, R.D.; Baker, S.S.; Gupta, A.; Holycross, M. Oxidant-induced disruption of intestinal epithelial barrier function: Role of protein tyrosine phosphorylation. Am. J. Physiol. 1997, 273, G812–G823.
- Rao, R.K.; Basuroy, S.; Rao, V.U.; Karnaky, K.J., Jr.; Gupta, A. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem. J. 2002, 368, 471–481.
- Sheth, P.; Seth, A.; Atkinson, K.J.; Gheyi, T.; Kale, G.; Giorgianni, F.; Desiderio, D.M.; Li, C.; Naren, A.; Rao, R. Acetaldehyde dissociates the PTP1B-E-cadherin-beta-catenin complex in Caco-2 cell monolayers by a phosphorylation-dependent mechanism. Biochem. J. 2007, 402, 291–300.
- Banan, A.; Choudhary, S.; Zhang, Y.; Fields, J.Z.; Keshavarzian, A. Ethanol-induced barrier dysfunction and its prevention by growth factors in human intestinal monolayers: Evidence for oxidative and cytoskeletal mechanisms. J. Pharmacol. Exp. Ther. 1999, 291, 1075–1085.
- Tang, Y.; Forsyth, C.B.; Farhadi, A.; Rangan, J.; Jakate, S.; Shaikh, M.; Banan, A.; Fields, J.Z.; Keshavarzian, A. Nitric oxide-mediated intestinal injury is required for alcohol-induced gut leakiness and liver damage. Alcohol. Clin. Exp. Res. 2009, 33, 1220–1230.
- Fukui, H. Role of Gut Dysbiosis in Liver Diseases: What Have We Learned So Far? Diseases 2019, 7, 58.
- Geirnaert, A.; Calatayud, M.; Grootaert, C.; Laukens, D.; Devriese, S.; Smagghe, G.; De Vos, M.; Boon, N.; Van de Wiele, T. Butyrate-producing bacteria supplemented in vitro to Crohn’s disease patient microbiota increased butyrate production and enhanced intestinal epithelial barrier integrity. Sci. Rep. 2017, 7, 11450.
- Bjørkhaug, S.T.; Aanes, H.; Neupane, S.P.; Bramness, J.G.; Malvik, S.; Henriksen, C.; Skar, V.; Medhus, A.W.; Valeur, J. Characterization of gut microbiota composition and functions in patients with chronic alcohol overconsumption. Gut Microbes 2019, 10, 663–675.
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol. Clin. Exp. Res. 2014, 38, 1489–1501.
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597.
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978.
- Yan, A.W.; Fouts, D.E.; Brandl, J.; Stärkel, P.; Torralba, M.; Schott, E.; Tsukamoto, H.; Nelson, K.E.; Brenner, D.A.; Schnabl, B. Enteric dysbiosis associated with a mouse model of alcoholic liver disease. Hepatology 2011, 53, 96–105.
- Bode, J.C.; Bode, C.; Heidelbach, R.; Dürr, H.K.; Martini, G.A. Jejunal microflora in patients with chronic alcohol abuse. Hepatogastroenterology 1984, 31, 30–34.
- Casafont Morencos, F.; de las Heras Castaño, G.; Martín Ramos, L.; López Arias, M.J.; Ledesma, F.; Pons Romero, F. Small bowel bacterial overgrowth in patients with alcoholic cirrhosis. Dig. Dis. Sci. 1996, 41, 552–556.
- Nanji, A.A.; Jokelainen, K.; Fotouhinia, M.; Rahemtulla, A.; Thomas, P.; Tipoe, G.L.; Su, G.L.; Dannenberg, A.J. Increased severity of alcoholic liver injury in female rats: Role of oxidative stress, endotoxin, and chemokines. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1348–G1356.
- Mathurin, P.; Deng, Q.G.; Keshavarzian, A.; Choudhary, S.; Holmes, E.W.; Tsukamoto, H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 2000, 32, 1008–1017.
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361.
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577.
- Ju, C.; Mandrekar, P. Macrophages and Alcohol-Related Liver Inflammation. Alcohol Res. 2015, 37, 251–262.
- Petrasek, J.; Iracheta-Vellve, A.; Saha, B.; Satishchandran, A.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Metabolic danger signals, uric acid and ATP, mediate inflammatory cross-talk between hepatocytes and immune cells in alcoholic liver disease. J. Leukoc. Biol. 2015, 98, 249–256.
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and ATP, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.Y.; Kurt-Jones, E.A.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489.
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404.
- Larson, S.R.; Atif, S.M.; Gibbings, S.L.; Thomas, S.M.; Prabagar, M.G.; Danhorn, T.; Leach, S.M.; Henson, P.M.; Jakubzick, C.V. Ly6C+ monocyte efferocytosis and cross-presentation of cell-associated antigens. Cell Death Differ. 2016, 23, 997–1003.
- Dou, L.; Shi, X.; He, X.; Gao, Y. Macrophage Phenotype and Function in Liver Disorder. Front. Immunol. 2019, 10, 3112.
- Badwey, J.A.; Karnovsky, M.L. Active oxygen species and the functions of phagocytic leukocytes. Annu. Rev. Biochem. 1980, 49, 695–726.
- Kono, H.; Rusyn, I.; Yin, M.; Gäbele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Investig. 2000, 106, 867–872.
- Thakur, V.; Pritchard, M.T.; McMullen, M.R.; Wang, Q.; Nagy, L.E. Chronic ethanol feeding increases activation of NADPH oxidase by lipopolysaccharide in rat Kupffer cells: Role of increased reactive oxygen in LPS-stimulated ERK1/2 activation and TNF-alpha production. J. Leukoc. Biol. 2006, 79, 1348–1356.
- Liaskou, E.; Klemsdal Henriksen, E.K.; Holm, K.; Kaveh, F.; Hamm, D.; Fear, J.; Viken, M.K.; Hov, J.R.; Melum, E.; Robins, H.; et al. High-throughput T-cell receptor sequencing across chronic liver diseases reveals distinct disease-associated repertoires. Hepatology 2016, 63, 1608–1619.
- Gao, B.; Ahmad, M.F.; Nagy, L.E.; Tsukamoto, H. Inflammatory pathways in alcoholic steatohepatitis. J. Hepatol. 2019, 70, 249–259.
- Chedid, A.; Mendenhall, C.L.; Moritz, T.E.; French, S.W.; Chen, T.S.; Morgan, T.R.; Roselle, G.A.; Nemchausky, B.A.; Tamburro, C.H.; Schiff, E.R.; et al. Cell-mediated hepatic injury in alcoholic liver disease. Veterans Affairs Cooperative Study Group 275. Gastroenterology 1993, 105, 254–266.
- Lin, F.; Taylor, N.J.; Su, H.; Huang, X.; Hussain, M.J.; Abeles, R.D.; Blackmore, L.; Zhou, Y.; Ikbal, M.M.; Heaton, N.; et al. Alcohol dehydrogenase-specific T-cell responses are associated with alcohol consumption in patients with alcohol-related cirrhosis. Hepatology 2013, 58, 314–324.
- Markwick, L.J.; Riva, A.; Ryan, J.M.; Cooksley, H.; Palma, E.; Tranah, T.H.; Manakkat Vijay, G.K.; Vergis, N.; Thursz, M.; Evans, A.; et al. Blockade of PD1 and TIM3 restores innate and adaptive immunity in patients with acute alcoholic hepatitis. Gastroenterology 2015, 148, 590–602.
- Støy, S.; Sandahl, T.D.; Dige, A.K.; Agnholt, J.; Rasmussen, T.K.; Grønbæk, H.; Deleuran, B.; Vilstrup, H. Highest frequencies of interleukin-22-producing T helper cells in alcoholic hepatitis patients with a favourable short-term course. PLoS ONE 2013, 8, e55101.
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785.
- Rutz, S.; Eidenschenk, C.; Ouyang, W. IL-22, not simply a Th17 cytokine. Immunol. Rev. 2013, 252, 116–132.
- Kumar, V. NKT-cell subsets: Promoters and protectors in inflammatory liver disease. J. Hepatol. 2013, 59, 618–620.
- Maricic, I.; Sheng, H.; Marrero, I.; Seki, E.; Kisseleva, T.; Chaturvedi, S.; Molle, N.; Mathews, S.A.; Gao, B.; Kumar, V. Inhibition of type I natural killer T cells by retinoids or following sulfatide-mediated activation of type II natural killer T cells attenuates alcoholic liver disease in mice. Hepatology 2015, 61, 1357–1369.
- Xiao, X.; Cai, J. Mucosal-Associated Invariant T Cells: New Insights into Antigen Recognition and Activation. Front. Immunol. 2017, 8, 1540.
- Kurioka, A.; Jahun, A.S.; Hannaway, R.F.; Walker, L.J.; Fergusson, J.R.; Sverremark-Ekström, E.; Corbett, A.J.; Ussher, J.E.; Willberg, C.B.; Klenerman, P. Shared and Distinct Phenotypes and Functions of Human CD161++ Vα7.2+ T Cell Subsets. Front. Immunol. 2017, 8, 1031.
- Kurioka, A.; Walker, L.J.; Klenerman, P.; Willberg, C.B. MAIT cells: New guardians of the liver. Clin. Transl. Immunol. 2016, 5, e98.
- Li, Y.; Huang, B.; Jiang, X.; Chen, W.; Zhang, J.; Wei, Y.; Chen, Y.; Lian, M.; Bian, Z.; Miao, Q.; et al. Mucosal-Associated Invariant T Cells Improve Nonalcoholic Fatty Liver Disease Through Regulating Macrophage Polarization. Front. Immunol. 2018, 9, 1994.
- Klenerman, P.; Hinks, T.S.C.; Ussher, J.E. Biological functions of MAIT cells in tissues. Mol. Immunol. 2021, 130, 154–158.
- Franciszkiewicz, K.; Salou, M.; Legoux, F.; Zhou, Q.; Cui, Y.; Bessoles, S.; Lantz, O. MHC class I-related molecule, MR1, and mucosal-associated invariant T cells. Immunol. Rev. 2016, 272, 120–138.
- Riva, A.; Patel, V.; Kurioka, A.; Jeffery, H.C.; Wright, G.; Tarff, S.; Shawcross, D.; Ryan, J.M.; Evans, A.; Azarian, S.; et al. Mucosa-associated invariant T cells link intestinal immunity with antibacterial immune defects in alcoholic liver disease. Gut 2018, 67, 918–930.
- Dolganiuc, A.; Petrasek, J.; Kodys, K.; Catalano, D.; Mandrekar, P.; Velayudham, A.; Szabo, G. MicroRNA expression profile in Lieber-DeCarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol. Clin. Exp. Res. 2009, 33, 1704–1710.
- Blaya, D.; Coll, M.; Rodrigo-Torres, D.; Vila-Casadesus, M.; Altamirano, J.; Llopis, M.; Graupera, I.; Perea, L.; Aguilar-Bravo, B.; Diaz, A.; et al. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut 2016, 65, 1535–1545.
- Hwang, S.; Yang, Y.M. Exosomal microRNAs as diagnostic and therapeutic biomarkers in non-malignant liver diseases. Arch. Pharm. Res. 2021, 44, 574–587.
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018, 9, 402.
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y. microRNAs are ligands of Toll-like receptors. RNA 2013, 19, 737–739.
- Massey, V.L.; Qin, L.; Cabezas, J.; Caballeria, J.; Sancho-Bru, P.; Bataller, R.; Crews, F.T. TLR7-let-7 Signaling Contributes to Ethanol-Induced Hepatic Inflammatory Response in Mice and in Alcoholic Hepatitis. Alcohol. Clin. Exp. Res. 2018, 42, 2107–2122.
- Fan, X.; Wu, J.; Poulsen, K.L.; Kim, A.; Wu, X.; Huang, E.; Miyata, T.; Sanz-Garcia, C.; Nagy, L.E. Identification of a MicroRNA-E3 Ubiquitin Ligase Regulatory Network for Hepatocyte Death in Alcohol-Associated Hepatitis. Hepatol. Commun. 2021, 5, 830–845.
- Bala, S.; Petrasek, J.; Mundkur, S.; Catalano, D.; Levin, I.; Ward, J.; Alao, H.; Kodys, K.; Szabo, G. Circulating microRNAs in exosomes indicate hepatocyte injury and inflammation in alcoholic, drug-induced, and inflammatory liver diseases. Hepatology 2012, 56, 1946–1957.
- Babuta, M.; Furi, I.; Bala, S.; Bukong, T.N.; Lowe, P.; Catalano, D.; Calenda, C.; Kodys, K.; Szabo, G. Dysregulated Autophagy and Lysosome Function Are Linked to Exosome Production by Micro-RNA 155 in Alcoholic Liver Disease. Hepatology 2019, 70, 2123–2141.
- Wang, X.; He, Y.; Mackowiak, B.; Gao, B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut 2021, 70, 784–795.
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387.
- Wang, W.; Zhong, G.Z.; Long, K.B.; Liu, Y.; Liu, Y.Q.; Xu, A.L. Silencing miR-181b-5p upregulates PIAS1 to repress oxidative stress and inflammatory response in rats with alcoholic fatty liver disease through inhibiting PRMT1. Int. Immunopharmacol. 2021, 101, 108151.
- Dong, X.; Liu, H.; Chen, F.; Li, D.; Zhao, Y. MiR-214 promotes the alcohol-induced oxidative stress via down-regulation of glutathione reductase and cytochrome P450 oxidoreductase in liver cells. Alcohol. Clin. Exp. Res. 2014, 38, 68–77.
- Kumar, S.; Rani, R.; Karns, R.; Gandhi, C.R. Augmenter of liver regeneration protein deficiency promotes hepatic steatosis by inducing oxidative stress and microRNA-540 expression. FASEB J. 2019, 33, 3825–3840.
- Luo, J.; Hou, Y.; Ma, W.; Xie, M.; Jin, Y.; Xu, L.; Li, C.; Wang, Y.; Chen, J.; Chen, W.; et al. A novel mechanism underlying alcohol dehydrogenase expression: Hsa-miR-148a-3p promotes ADH4 expression via an AGO1-dependent manner in control and ethanol-exposed hepatic cells. Biochem. Pharmacol. 2021, 189, 114458.
- Luo, J.; Xie, M.; Hou, Y.; Ma, W.; Jin, Y.; Chen, J.; Li, C.; Zhao, K.; Chen, N.; Xu, L.; et al. A novel epigenetic mechanism unravels hsa-miR-148a-3p-mediated CYP2B6 downregulation in alcoholic hepatitis disease. Biochem. Pharmacol. 2021, 188, 114582.
- Fu, R.; Zhou, J.; Wang, R.; Sun, R.; Feng, D.; Wang, Z.; Zhao, Y.; Lv, L.; Tian, X.; Yao, J. Protocatechuic Acid-Mediated miR-219a-5p Activation Inhibits the p66shc Oxidant Pathway to Alleviate Alcoholic Liver Injury. Oxid. Med. Cell. Longev. 2019, 2019, 3527809.
- Khoruts, A.; Stahnke, L.; McClain, C.J.; Logan, G.; Allen, J.I. Circulating tumor necrosis factor, interleukin-1 and interleukin-6 concentrations in chronic alcoholic patients. Hepatology 1991, 13, 267–276.
- Ren, R.; He, Y.; Ding, D.; Cui, A.; Bao, H.; Ma, J.; Hou, X.; Li, Y.; Feng, D.; Li, X.; et al. Aging exaggerates acute-on-chronic alcohol-induced liver injury in mice and humans by inhibiting neutrophilic sirtuin 1-C/EBPalpha-miRNA-223 axis. Hepatology 2021.

