 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rances Blanco | + 4394 word(s) | 4394 | 2020-08-27 06:00:54 | | | |
| 2 | Felix Wu | -2482 word(s) | 1912 | 2020-11-04 03:04:10 | | |
Video Upload Options
High-risk human papillomavirus (HR-HPV) is etiologically associated with the development and progression of cervical cancer, although other factors are involved. Overall, reports suggest a potential link of EBV to the development of cervical carcinomas in two possible pathways: (1) Infecting epithelial cells, thus synergizing with HR-HPV (direct pathway), and/or (2) infecting tissue-infiltrating lymphocytes that could generate local immunosuppression (indirect pathway). However, further studies are needed for a better understanding of the EBV/HR-HPV coinfection role in cervical carcinogenesis, in which in situ hybridization (ISH) and/or immunohistochemical methods are mandatory for discriminating the cell type infected by EBV.
1. Introduction
The most important risk factor for cervical cancer development is infection with human papillomavirus (HPV) [1], with 99.7% of cervical carcinomas worldwide caused by high risk (HR)-HPV types, such as HPV16 or HPV18 [2][3]. While vaccines exist that protect against oncogenic HPV infection, global disparities still remain due to high costs [4][5]. However, HR-HPV infection is insufficient for cervical cancer development since low-grade squamous intraepithelial lesions (LSIL) usually regress to normal cells in cervical cytology or atypical squamous cells of undetermined significance (ASCUS). Only 3.6% of LSILs progress to high-grade squamous intraepithelial lesion (HSIL) [6], with a report of 50% regression in LSILs associated with HPV16 or HPV18 [7]. Accordingly, other host or environmental factors are necessary for cervical lesion progression, such as tobacco smoking (TS), oral contraceptive pills or immunosuppression, in particular infection with human immunodeficiency virus (HIV).
2. EBV Replication and Role in Cancer
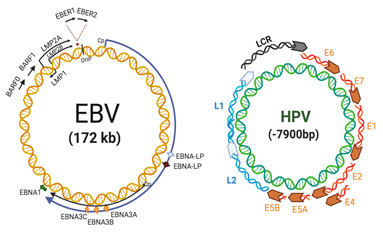
EBV, also known as human herpesvirus 4 (HHV-4) is a gammaherpervirus that infects more than 90% of the human population worldwide [8]. It is characterized by a double-stranded DNA genome of 172 kb in length, surrounded by an envelope carrying various surface glycoproteins, with tegument proteins filling the space between the membrane and the inner icosahedral capsid [9]. EBV is related to some B cell-derived malignancies such as Burkitt’s lymphoma (BL) and Hodgkin’s disease (HD). Importantly, EBV is associated with some epithelial tumors, including undifferentiated nasopharyngeal carcinomas (NPCs) and a subset of gastric carcinomas (GCs) [8]. Figure 1 (left) shows the EBV genome organization with locations of latent genes.
The mechanisms of EBV-mediated B-cell cancers are well known [10], although they are less understood in epithelial cells. In NPC cells, Epstein-Barr nuclear antigen 1 (EBNA1) has a key role in EBV persistency, decreasing p53 accumulation in response to DNA damage [11], whilst inducing epithelial mesenchymal transition (EMT), deregulating some related genes [12] and enhancing angiogenesis in vitro [13]. Latent membrane protein 1 (LMP1), another latent protein, impairs the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [14], activator protein 1 (AP-1) [15] and the Janus kinase/signal transducers and activators of transcription (JAK/STAT) [16] signaling pathways, leading to cell cycle disruption. Additionally, EBV-related epithelial tumors express BamHI-A rightward frame 1 (BARF1) [17], encoding a 220 amino acid lytic protein, secreted by EBV-infected epithelial cells as a soluble hexameric molecule (sBARF1) [18]. This viral protein hijacks human colony-stimulating factor 1 (hCSF-1), interfering with monocyte maturation [19] and impairing host immune responses against viral infections. It has been reported that BARF1 upregulates the expression of RelA and cyclin D1 and is able to reduce the cell cycle inhibitor p21WAF1 [20][21], promoting cell proliferation. Moreover, BARF1 activates the extracellular signal-regulated protein kinases 1 and 2 (ERK1/2)/c-Jun pathway [22] inhibiting apoptosis whilst increasing Bcl-2 and reducing both caspases and Bax [23]. Additionally, it inhibits interferon-alpha (IFN-α) production and release by mononuclear cells [24], impairing host immune responses. Interestingly, telomerase activation is observed in HPV-positive cervical cancer cells (HeLa) transfected with the BARF1 gene [25]. As BARF1 is expressed in EBV-associated NPC and GCs and not in lymphomas, BARF1 is considered an exclusive epithelial oncoprotein [25][26].
3. HPV in Cervical Cancer
HPV is a non-enveloped and exclusively intraepithelial virus, which comprises an 8 kb double-stranded circular DNA containing eight protein-coding genes divided into three major regions: Early, late and a noncoding region, known as the long control region (LCR). Early gene encoding non-structural proteins (E1 to E7) are involved in viral replication, transcription and transformation, while late transcribed genes encode for structural L1 and L2 proteins [27]. More than 210 HPV types have been identified by L1 sequencing, classified in HR-HPV (e.g., 16, 18 and 31) and low-risk (LR)-HPV types (e.g., 6, 7 and 11) according to the oncogenic potential [28][29]. Figure 1 (right) shows the HPV genome organization.
HR-HPV infection is etiologically associated with cervical carcinomas, anogenital and a subset of head and neck squamous cell carcinomas (HNSCCs). Integration of HR-HPV genomes into the host is an important hit, although not a requisite for epithelial carcinogenesis [30][31]. This is common in HSIL and cervical SCC compared with normal or LSIL tissues[32]. Moreover, HR-HPV (e.g., 16 and 18) integration in HSIL is frequently accompanied by chromosomal abnormalities [33], with E2 region loss during integration, leading to constitutive E6 and E7 protein expression [34]. E6 is a ~150 amino acid protein comprising two zinc finger binding domains connected by a 36 amino acid long-linker, and the carboxy terminal domain containing a PDZ-binding motif interacting with cell proteins [35]. E6 promotes loss of p53 via E6-associated protein (E6-AP)-mediated ubiquitination and proteasome degradation, inhibits apoptosis [36] and activates Mitogen-activated protein kinase (MAPK) and the mechanistic target of rapamycin complex 1 (mTORC1) pathways [37][38]. Additionally, E6 targets the pro-apoptotic proteins Bak for degradation [39] and others such as the Fas-associated protein with death domain (FADD) and caspase-8, disrupting the apoptotic program [40]. Likewise, E7 is a 100 amino acid protein comprising three conserved regions denoted CR1, CR2 and CR3, with CR3 containing two CXXC motifs separated by 29 or 30 residues. Moreover, the E7 carboxyl terminal domain contains a zinc-binding motif mediating interaction with cellular proteins [41]. E7 targets retinoblastoma protein (pRb) for ubiquitination, leading to E2F transcription factor release, allowing cell entry to S-phase [42]. Both E6 and E7 interact with c-Myc inducing human telomerase reverse transcriptase (hTERT) promoter activation and hTERT expression, enabling cell evasion of senescence [43][44]. E6 and E7 also upregulate expression of EMT markers such as N-cadherin, Fibronectin and Vimentin, increasing cell migration and invasiveness [45]. Moreover, E6 and E7 inhibit interferon (IFN) antiviral activity and decrease tumor necrosis factor alpha (TNF-α) and (interleukin-1 beta) IL-1β secretion by macrophages, enabling immune evasion [46].
HR-HPV infection is insufficient for cervical carcinogenesis and involvement of other cofactors is essential. While factors involved in HR-HPV integration into the host genome are unclear [47], a prerequisite is DNA damage [48], TS being a very important factor for cervical cancer development [49] by activating the HPV16 p97 promoter and leading to E6 and E7 overexpression [50]. Moreover, HPV16 E6 and E7 collaborate with TS, increasing the tumor properties of epithelial cells [51][52], although other potential cofactors such as chronic inflammation and bacterial or viral coinfection are also suspected.
4. Mechanisms of HPV/EBV-Mediated Cervical Carcinogenesis
Experimental approaches evaluating molecular mechanisms involved in EBV/HR-HPV coinfection are limited. However, EBV LMP1 in combination with HPV16 E6 viral proteins in transformed mouse embryonic fibroblasts (MEFs) reduces components of DNA damage response (DDR) such as p53, pRb and p27, whilst increasing checkpoint kinase 1 (CHK1), NF-κB signaling, v-akt murine thymoma viral oncogene (Akt) and MAPK signaling [53][54]. In addition, LMP1 induces down-regulation of E-cadherin expression [55] and also regulates TWIST [56] and SNAIL [57] transcription factors and others related with cell motility. Moreover, LMP1 and E6 co-expression induces cell proliferation, resistance to apoptosis, anchorage-independent growth and tumor-formation ability in nude mice compared with single expression of EBNA1 or E6 [36]. In EBV-infected NPC cells, EBNA1 plays a role in EMT through inhibition of both microRNA (miR)-200a and -200b expression, while it up-regulates the expression of ZEB1 and ZEB2, their target genes [12]. Interestingly, double expression of LMP1 and HR-HPV E6 relates with a more aggressive form of malignant tumor such as breast adenocarcinoma [58] and cervical SCC [59]. Thereby, in the case of EBV-associated GCs, EBNA1 may induce constitutive and also IFNγ-inducible programmed death-ligand 1 (PD-L1) expression in EBV-infected epithelial- cells [60]. In HPV+ HeLa and SiHa cervical cells, transfection with a small interfering RNA (siRNA) for Myc knock down was able to reduce the hTERT promoter activity [61]. HPV16 E6 activates hTERT gene transcription through induction of c-Myc, which is overexpressed in cervical carcinomas [62][63]. Moreover, hTERT activation in BARF1-transfected PATAS monkey kidney cells was accompanied by up-regulation of c-Myc, while in HeLa cells BARF1 induces activation of telomerase binding directly to initiator elements in the hTERT promoter region [25]. Alternatively, both HPV18 E6 and E7 oncoproteins are necessary to increase EBV genome maintenance in normal oral keratinocytes (NOKs) and to induce the reactivation of the EBV lytic program in suprabasal layers of oral epithelial from an in vitro organotypic raft culture model [64]. However, a significant reduction in EBV immediate early (BZLF1, BRLF1) and early (BALF5, BMRF1) gene expression with increased expression of EBER1 was evidenced in HPV+/EBV+ human foreskin keratinocytes (HFK) compared to that in HPV-/EBV+ rafts [65]. In cervical cells, increased levels of EBER1 may contribute to the transition from inflammation to oncogenesis of HPV-associated cervical cancer by modulating innate immune responses [66]. In addition, LMP1 reduces the presence of HPV16 with no expression change in the EBNA1 and EBNA2 latent genes, suggesting that EBV latency is favored over lytic replication in HPV16+ cells. However, LMP1 is mostly expressed in B-cell lymphomas, lymphoproliferative disorders and NPC [67], but less in cervical and GCs [68][69]. In uterine cervix, latency III EBV infection is increased in CIN and cervical SCC compared with non-malignant samples [68], conferring a long-term persistence of EBV infection in malignant cells. Furthermore, reduced EBV replication mediated by HPV16 E7 is related to retarded expression of some markers related with early epithelial cell differentiation [65]. Similarly, cervical SCC EBV/HR-HPV coinfection is associated with decreased cell differentiation [59], closely related with a more aggressive tumor. Additionally, NOKs and FaDu hypopharyngeal carcinoma cells coinfected with EBV and HR-HPV show increased invasiveness in the lysophosphatidic acid (LPA) presence (glycerophospholipid able to stimulate cell migration) compared to EBV-/HR-HPV- and EBV+/HR-HPV- cells [70], resembling the effects of coinfection in cervical SCC [59]. According to our knowledge, the levels of BARF1 in cervical cancer have been examined in only one study, which reported 27% detection in tumors from Algerian women [71]. As previously stated, this protein has been suggested as an epithelial EBV oncogene, which is expressed in a cell differentiation-dependent manner [72]. Due to this lytic protein being expressed in a majority of EBV+ NPC and GC cells, the possibility that this protein is involved in EBV+ cervical carcinomas, working in both oncogenic process and immune evasion is plausible. Considering that EBV infection in normal epithelial cells is exclusively lytic [73][74], an interesting point is the establishment of EBV latency in epithelial cervical cells. In this respect, it has been suggested that previous DNA damage in cells is a requisite [75]. Thus, we can propose a scenario in which previous HPV infected and subsequently DNA-damaged cells (e.g., by HPV E7) are particularly susceptible to EBV latency establishment. Moreover, we can speculate on the possibility that HPV is involved in the activation of the EBV abortive lytic cycle, promoting expression of some lytic genes, such as Zebra or Rta, which has been reported to be important in EBV-mediated carcinogenesis. A hypothetical model in which HPV infection favors EBV latency establishment in epithelial cervical cancer cells is shown in Figure 2.
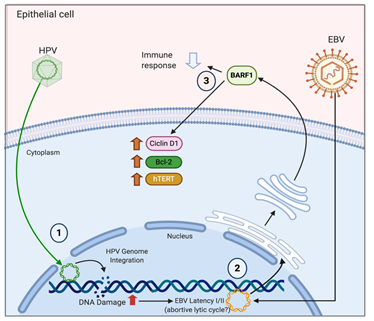
Figure 2. A hypothetical model of HR-HPV/EBV interaction in cervical epithelial cells. 1. HR-HPV infection and viral integration promotes DNA damage and genomic instability in cervical cells; 2. DNA damage promotes the establishment of EBV latency (I/II) with potential expression of some lytic genes such as BARF1 (abortive lytic expression); 3. BARF1 (and other viral oncogenes) promotes oncogenic changes and immune evasion, cooperating with HR-HPV. Created by BioRender.com.
References
- Walboomers, J.M.; Jacobs, M.V.; Manos, M.M.; Bosch, F.X.; Kummer, J.A.; Shah, K.V.; Snijders, P.J.; Peto, J.; Meijer, C.J.; Munoz, N. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. Pathol.1999, 189, 12–19.
- Ciapponi, A.; Bardach, A.; Glujovsky, D.; Gibbons, L.; Piconni, M.A. Type-specific HPV prevalence in cervical cancer and high-grade lesions in Latin America and the Caribbean: Systematic review and meta-analysis. PLoS ONE 2011, 6, e25493.
- Bernard, E.; Pons-Salort, M.; Favre, M.; Heard, I.; Delarocque-Astagneau, E.; Guillemot, D.; Thiebaut, A.C.M. Comparing human papillomavirus prevalences in women with normal cytology or invasive cervical cancer to rank genotypes according to their oncogenic potential: A meta-analysis of observational studies. BMC Infect. Dis. 2013, 13, 373.
- Howard, N.; Gallagher, K.E.; Mounier-Jack, S.; Burchett, H.E.D.; Kabakama, S.; Lamontagne, D.S.; Watson-Jones, D. What works for human papillomavirus vaccine introduction in low and middle-income countries? Papillomavirus Res. (Amst. Neth.) 2017, 4, 22–25.
- Murillo, R.; Reyes, C.O. Human papillomavirus (HPV) vaccination: From clinical studies to immunization programs. Int. J. Gynecol. Cancer 2019, 29, 1317–1326.
- Schlecht, N.F.; Platt, R.W.; Duarte-Franco, E.; Costa, M.C.; Sobrinho, J.P.; Prado, J.C.M.; Ferenczy, A.; Rohan, T.E.; Villa, L.L.; Franco, E. Human papillomavirus infection and time to progression and regression of cervical intraepithelial neoplasia. J. Natl. Cancer Inst. 2003, 95, 1336–1343.
- Silveira, F.; Almeida, G.; Furtado, Y.; Cavalcanti, S.M.B.; Silva, K.; Maldonado, P.; Carvalho, M. The association of HPV genotype with the regression, persistence or progression of low-grade squamous intraepithelial lesions. Exp. Mol. Pathol. 2015, 99, 702–706.
- Farrell, P.J. Epstein-Barr virus and cancer. Annu. Rev. Pathol. 2019, 14, 29–53.
- Grünewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398.
- Grywalska, E.; Rolinski, J. Epstein-Barr virus-associated lymphomas. Semin. Oncol. 2015, 42, 291–303.
- Sivachandran, N.; Wang, X.; Frappier, L. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J. Virol. 2012, 86, 6146–6158.
- Wang, L.; Tian, W.-D.; Xu, X.; Nie, B.; Lu, J.; Liu, X.; Zhang, B.; Dong, Q.; Sunwoo, J.B.; Li, G.; et al. Epstein-Barr virus nuclear antigen 1 (EBNA1) protein induction of epithelial-mesenchymal transition in nasopharyngeal carcinoma cells. Cancer 2013, 120, 363–372.
- O’Neil, J.D.; Owen, T.J.; Wood, V.H.J.; Date, K.L.; Valentine, R.; Chukwuma, M.B.; Arrand, J.R.; Dawson, C.W.; Young, L.S. Epstein-Barr virus-encoded EBNA1 modulates the AP-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J. Gen. Virol. 2008, 89, 2833–2842.
- Yin, L.; Liao, W.; Deng, X.; Tang, M.; Gu, H.; Li, X.; Yi, W.; Cao, Y. LMP1 activates NF-kappa B via degradation of I kappa B alpha in nasopharyngeal carcinoma cells. Chin. Med. J. 2001, 114, 718–722.
- Yang, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y.; Song, X. Heterodimer formation between c-Jun and Jun B proteins mediated by Epstein-Barr virus encoded latent membrane protein 1. Cell Sign. 2004, 16, 1153–1162.
- Chen, H.; Lee, J.; Zong, Y.; Borowitz, M.; Ng, M.H.; Ambinder, R.F.; Hayward, S.D. Linkage between STAT regulation and Epstein-Barr virus gene expression in tumors. J. Virol. 2001, 75, 2929–2937.
- Seto, E.; Yang, L.; Middeldorp, J.M.; Sheen, T.-S.; Chen, J.-Y.; Fukayama, M.; Eizuru, Y.; Ooka, T.; Takada, K. Epstein-Barr virus (EBV)-encodedBARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 2005, 76, 82–88.
- Hoebe, E.K.; Le Large, T.Y.S.; Greijer, A.E.; Middeldorp, J.M. BamHI-A rightward frame 1, an Epstein-Barr virus-encoded oncogene and immune modulator. Rev. Med. Virol. 2013, 23, 367–383.
- Elegheert, J.; Bracke, N.; Pouliot, P.; Gutsche, I.; Shkumatov, A.V.; Tarbouriech, N.; Verstraete, K.; Bekaert, A.; Burmeister, W.P.; Svergun, D.I.; et al. Allosteric competitive inactivation of hematopoietic CSF-1 signaling by the viral decoy receptor BARF1. Nat. Struct. Mol. Boil. 2012, 19, 938–947.
- Wiech, T.; Nikolopoulos, E.; Lassman, S.; Heidt, T.; Schöpflin, A.; Sarbia, M.; Werner, M.; Shimizu, Y.; Sakka, E.; Ooka, T.; et al. Cyclin D1 expression is induced by viral BARF1 and is overexpressed in EBV-associated gastric cancer. Virchows Arch. 2008, 452, 621–627.
- Chang, M.S.; Kim, D.H.; Roh, J.K.; Middeldorp, J.M.; Kim, Y.S.; Kim, S.; Han, S.; Kim, C.W.; Lee, B.L.; Kim, W.H.; et al. Epstein-Barr Virus-Encoded BARF1 Promotes Proliferation of Gastric Carcinoma Cells through Regulation of NF-B. J. Virol. 2013, 87, 10515–10523.
- Zhang, Y.; Xu, M.; Zhang, X.; Chu, F.; Zhou, T. MAPK/c-Jun signaling pathway contributes to the upregulation of the anti-apoptotic proteins Bcl-2 and Bcl-xL induced by Epstein-Barr virus-encoded BARF1 in gastric carcinoma cells. Oncol. Lett. 2018, 15, 7537–7544.
- Zhang, Y.; Ohyashiki, J.H.; Takaku, T.; Shimizu, N.; Ohyashiki, K. Transcriptional profiling of Epstein-Barr virus (EBV) genes and host cellular genes in nasal NK/T-cell lymphoma and chronic active EBV infection. Br. J. Cancer 2006, 94, 599–608.
- Cohen, J.I.; Lekstrom, K. Epstein-Barr virus BARF1 protein is dispensable for B-cell transformation and inhibits alpha interferon secretion from mononuclear cells. J. Virol. 1999, 73, 7627–7632.
- Jiang, R.; Cabras, G.; Sheng, W.; Zeng, Y.; Ooka, T. Synergism of BARF1 with ras induces malignant transformation in primary primate epithelial cells and human nasopharyngeal epithelial cells. Neoplasia 2009, 11, 964–973.
- Hayes, D.P.; Brink, A.A.; Vervoort, M.B.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Expression of Epstein-Barr virus (EBV) transcripts encoding homologues to important human proteins in diverse EBV associated diseases. Mol. Pathol. 1999, 52, 97–103.
- Pinidis, P.; Tsikouras, P.; Iatrakis, G.; Zervoudis, S.; Koukouli, Z.; Bothou, A.; Galazios, G.; Vladareanu, S. Human papilloma virus’ life cycle and carcinogenesis. Maedica (Buchar.) 2016, 11, 48–54.
- Muñoz, N.; Bosch, F.X.; de Sanjosé, S.; Herrero, R.; Castellsagué, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J.; Group, I.A.f.R.o.C.M.C.C.S. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527.
- De Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27.
- Park, J.S.; Hwang, E.S.; Park, S.N.; Ahn, H.K.; Um, S.J.; Kim, C.J.; Kim, S.J.; Namkoong, S.E. Physical status and expression of hpv genes in cervical cancers. Gynecol. Oncol. 1997, 65, 121–129.
- Li, K.; Jin, X.; Fang, Y.; Wang, C.; Gong, M.; Chen, P.; Liu, J.; Deng, D.; Ai, J. Correlation between physical status of human papilloma virus and cervical carcinogenesis. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 97–102.
- Klaes, R.; Woerner, S.M.; Ridder, R.; Wentzensen, N.; Duerst, M.; Schneider, A.; Lotz, B.; Melsheimer, P.; von Knebel Doeberitz, M. Detection of high-risk cervical intraepithelial neoplasia and cervical cancer by amplification of transcripts derived from integrated papillomavirus oncogenes. Cancer Res. 1999, 59, 6132–6136.
- Hopman, A.H.; Smedts, F.; Dignef, W.; Ummelen, M.; Sonke, G.; Mravunac, M.; Vooijs, G.P.; Speel, E.J.; Ramaekers, F.C. Transition of high-grade cervical intraepithelial neoplasia to micro-invasive carcinoma is characterized by integration of HPV 16/18 and numerical chromosome abnormalities. J. Pathol. 2004, 202, 23–33.
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460.
- Thomas, M.; Laura, R.; Hepner, K.; Guccione, E.; Sawyers, C.; Lasky, L.; Banks, L. Oncogenic human papillomavirus E6 proteins target the MAGI-2 and MAGI-3 proteins for degradation. Oncogene 2002, 21, 5088–5096.
- Pflaum, J.; Schlosser, S.; Müller, M. p53 family and cellular stress responses in cancer. Front. Oncol. 2014, 4, 285.
- Chakrabarti, O.; Veeraraghavalu, K.; Tergaonkar, V.; Liu, Y.; Androphy, E.J.; Stanley, M.A.; Krishna, S. Human papillomavirus type 16 E6 amino acid 83 variants enhance E6-mediated MAPK signaling and differentially regulate tumorigenesis by notch signaling and oncogenic Ras. J. Virol. 2004, 78, 5934–5945.
- Spangle, J.M.; Münger, K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. J. Virol. 2010, 84, 9398–9407.
- Garnett, T.O.; Duerksen-Hughes, P.J. Modulation of apoptosis by human papillomavirus (HPV) oncoproteins. Arch Virol. 2006, 151, 2321–2335.
- Yuan, C.H.; Filippova, M.; Tungteakkhun, S.S.; Duerksen-Hughes, P.J.; Krstenansky, J.L. Small molecule inhibitors of the HPV16-E6 interaction with caspase 8. Bioorg. Med. Chem. Lett. 2012, 22, 2125–2129.
- Ohlenschläger, O.; Seiboth, T.; Zengerling, H.; Briese, L.; Marchanka, A.; Ramachandran, R.; Baum, M.; Korbas, M.; Meyer-Klaucke, W.; Dürst, M.; et al. Solution structure of the partially folded high-risk human papilloma virus 45 oncoprotein E7. Oncogene 2006, 25, 5953–5959.
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res 1996, 56, 4620–4624.
- Veldman, T.; Liu, X.; Yuan, H.; Schlegel, R. Human papillomavirus E6 and Myc proteins associate in vivo and bind to and cooperatively activate the telomerase reverse transcriptase promoter. Proc. Natl. Acad. Sci. USA 2003, 100, 8211–8216.
- Wang, Y.W.; Chang, H.S.; Lin, C.H.; Yu, W.C. HPV-18 E7 conjugates to c-Myc and mediates its transcriptional activity. Int. J. Biochem. Cell Boil. 2007, 39, 402–412.
- Hellner, K.; Mar, J.; Fang, F.; Quackenbush, J.; Münger, K. HPV16 E7 oncogene expression in normal human epithelial cells causes molecular changes indicative of an epithelial to mesenchymal transition. Virology 2009, 391, 57–63.
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186.
- Oyervides-Muñoz, M.A.; Pérez-Maya, A.A.; Rodríguez-Gutiérrez, H.F.; Gómez-Macias, G.S.; Fajardo-Ramírez, O.R.; Treviño, V.; Barrera-Saldaña, H.A.; Garza-Rodríguez, M.L. Understanding the HPV integration and its progression to cervical cancer. Infect. Genet. Evol. 2018, 61, 134–144.
- Williams, V.M.; Filippova, M.; Soto, U.; Duerksen-Hughes, P.J. HPV-DNA integration and carcinogenesis: Putative roles for inflammation and oxidative stress. Future Virol. 2011, 6, 45–57.
- Deacon, J.M.; Evans, C.D.; Yule, R.; Desai, M.; Binns, W.; Taylor, C.; Peto, J. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: A case–control study nested within the Manchester cohort. Br. J. Cancer 2000, 83, 1565–1572.
- Peña, N.; Carrillo, D.; Muñoz, J.P.; Chnaiderman, J.; Urzúa, U.; León, O.; Tornesello, M.L.; Corvalán, A.H.; Soto-Rifo, R.; Aguayo, F. Tobacco smoke activates human papillomavirus 16 p97 promoter and cooperates with high-risk E6/E7 for oxidative DNA damage in lung cells. PLoS ONE 2015, 10, e0123029.
- Muñoz, J.P.; González, C.; Parra, B.; Corvalán, A.H.; Tornesello, M.L.; Eizuru, Y.; Aguayo, F. Functional interaction between human papillomavirus type 16 E6 and E7 oncoproteins and cigarette smoke components in lung epithelial cells. PLoS ONE 2012, 7, e38178.
- Muñoz, J.P.; Carrillo-Beltrán, D.; Aedo-Aguilera, V.; Calaf, G.M.; León, O.; Maldonado, E.; Tapia, J.C.; Boccardo, E.; Ozbun, M.A.; Aguayo, F. Tobacco exposure enhances human papillomavirus 16 oncogene expression via EGFR/PI3K/Akt/c-Jun signaling pathway in cervical cancer cells. Front. Microbiol. 2018, 9, 3022.
- Shimabuku, T.; Tamanaha, A.; Kitamura, B.; Tanabe, Y.; Tawata, N.; Ikehara, F.; Arakaki, K.; Kinjo, T. Dual expression of Epstein-Barr virus, latent membrane protein-1 and human papillomavirus-16 E6 transform primary mouse embryonic fibroblasts through NF-κB signaling. Int. J. Clin. Exp. Pathol. 2014, 7, 1920–1934.
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, 21.
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 involves c-Jun NH2-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676.
- Horikawa, T.; Yang, J.; Kondo, S.; Yoshizaki, T.; Joab, I.; Furukawa, M.; Pagano, J.S. Twist and epithelial-mesenchymal transition are induced by the EBV oncoprotein latent membrane protein 1 and are Associated with metastatic nasopharyngeal carcinoma. Cancer Res. 2007, 67, 1970–1978.
- Horikawa, T.; Yoshizaki, T.; Kondo, S.; Furukawa, M.; Kaizaki, Y.; Pagano, J.S. Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial–mesenchymal transition in metastatic nasopharyngeal carcinoma. Br. J. Cancer 2011, 104, 1160–1167.
- Al Moustafa, A.E.; Al-Antary, N.; Aboulkassim, T.; Akil, N.; Batist, G.; Yasmeen, A. Co-prevalence of Epstein-Barr virus and high-risk human papillomaviruses in Syrian women with breast cancer. Vaccines Immunother.2016, 12, 1936–1939.
- Al-Thawadi, H.; Ghabreau, L.; Aboulkassim, T.; Yasmeen, A.; Vranic, S.; Batist, G.; Al Moustafa, A.E. Co-Incidence of Epstein-Barr Virus and High-Risk Human Papillomaviruses in Cervical Cancer of Syrian W omen. Oncol.2018, 8, 250.
- Moon, J.W.; Kong, S.K.; Kim, B.S.; Kim, H.J.; Lim, H.; Noh, K.; Kim, Y.; Choi, J.W.; Lee, J.H.; Kim, Y.S. IFNγ induces PD-L1 overexpression by JAK2/STAT1/IRF-1 signaling in EBV-positive gastric carcinoma. Sci. Rep. 2017, 7, 17810.
- Zhang, Y.; Dakic, A.; Chen, R.; Dai, Y.; Schlegel, R.; Liu, X. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget 2017, 8, 96323–96339.
- McMurray, H.R.; McCance, D.J. Human papillomavirus type 16 E6 activates TERT gene transcription through induction of c-Myc and release of USF-mediated repression. J. Virol. 2003, 77, 9852–9861.
- Zhang, A.; Månér, S.; Betz, R.; Angström, T.; Stendahl, U.; Bergman, F.; Zetterberg, A.; Wallin, K.L. Genetic alterations in cervical carcinomas: Frequent low-level amplifications of oncogenes are associated with human papillomavirus infection. Int. J. Cancer 2002, 101, 427–433.
- Makielski, K.R.; Lee, D.; Lorenz, L.D.; Nawandar, D.M.; Chiu, Y.F.; Kenney, S.C.; Lambert, P.F. Human papillomavirus promotes Epstein-Barr virus maintenance and lytic reactivation in immortalized oral keratinocytes. Virology 2016, 495, 52–62.
- Guidry, J.T.; Myers, J.E.; Bienkowska-Haba, M.; Songock, W.K.; Ma, X.; Shi, M.; Nathan, C.O.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Inhibition of Epstein-Barr Virus Replication in Human Papillomavirus-Immortalized Keratinocytes. J. Virol. 2018, 93, 2.
- Aromseree, S.; Middeldorp, J.M.; Pientong, C.; van Eijndhoven, M.; Ramayanti, O.; Lougheed, S.M.; Pegtel, D.M.; Steenbergen, R.D.; Ekalaksananan, T. High Levels of EBV-Encoded RNA 1 (EBER1) Trigger Interferon and Inflammation-Related Genes in Keratinocytes Expressing HPV16 E6/E7. PLoS ONE 2017, 12, e0169290.
- Dugan, J.P.; Coleman, B.P.; Haverkos, B. Opportunities to target the life cycle of epstein-barr virus (EBV) in EBV-associated lymphoproliferative disorders. Oncol.2019, 9, 127.
- Sasagawa, T.; Shimakage, M.; Nakamura, M.; Sakaike, J.; Ishikawa, H.; Inoue, M. Epstein-Barr virus (EBV) genes expression in cervical intraepithelial neoplasia and invasive cervical cancer: A comparative study with human papillomavirus (HPV) infection. Pathol.2000, 31, 318–326.
- Wang, A.; Zhang, W.; Jin, M.; Zhang, J.; Li, S.; Tong, F.; Zhou, Y. Differential expression of EBV proteins LMP1 and BHFR1 in EBV-associated gastric and nasopharyngeal cancer tissues. Med. Rep.2016, 13, 4151–4158.
- Jiang, R.; Ekshyyan, O.; Moore-Medlin, T.; Rong, X.; Nathan, S.; Gu, X.; Abreo, F.; Rosenthal, E.L.; Shi, M.; Guidry, J.T.; et al. Association between human papilloma virus/Epstein-Barr virus coinfection and oral carcinogenesis. Oral Pathol. Med.2014, 44, 28–36.
- Khenchouche, A.; Sadouki, N.; Boudriche, A.; Houali, K.; Graba, A.; Ooka, T.; Bouguermouh, A. Human Papillomavirus and Epstein-Barr virus co-infection in Cervical Carcinoma in Algerian women. J.2013, 10, 340.
- Hoebe, E.; Wille, C.; Hagemeier, S.; Kenney, S.; Greijer, A.; Middeldorp, J. Epstein-Barr Virus Gene BARF1 expression is regulated by the epithelial differentiation factor ΔNp63α in undifferentiated nasopharyngeal carcinoma. Cancers (Basel) 2018, 10, 76.
- Temple, R.M.; Meyers, C.; Sample, C.E. Generation and infection of organotypic cultures with Epstein-Barr virus. Methods Mol. Biol. 2017, 1532, 65–78.
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein-Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549.
- Tsang, C.M.; Deng, W.; Yip, Y.L.; Zeng, M.S.; Lo, K.W.; Tsao, S.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555.


