 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | David J. Robbins | + 2028 word(s) | 2028 | 2020-08-19 05:45:01 | | | |
| 2 | Bruce Ren | Meta information modification | 2028 | 2020-10-26 10:56:53 | | |
Video Upload Options
1. Introduction
The evolutionarily conserved Wnt signaling cascade has been extensively studied for over three decades and has been shown to regulate numerous cellular events during development and adult tissue homeostasis, as well as in disease when deregulated [1][2]. The term ‘Wnt’ was first coined in 1991 from a combination of wingless, the gene that patterns the development of many tissues in Drosophila melanogaster, including the wing, and its mouse ortholog Int-1, the proto-oncogene that regulates mammary tumorigenesis in mice [3]. Wnt ligands are a family of secreted glycoproteins that trigger a unique signal transduction network [4][5]. Wnt proteins undergo palmitoylation by the membrane-bound O-acyltransferase porcupine (PORCN) in the endoplasmic reticulum (ER) [6][7][8]. This modification promotes Wnt export from the ER and subsequently out of the cell and facilitates its activation and binding to the membrane receptor frizzled (Fzd) [6][7][8][9][10]. Two distinct arms of the Wnt signaling network have been identified, defined by their dependence on β-catenin: canonical Wnt/β-catenin signaling and non-canonical Wnt signaling. In this review, we focus on the canonical Wnt signaling pathway, discussing the role of its various critical components in cancer, with a focus on the established negative regulator of Wnt signaling, casein kinase 1α (CK1α).
2. CK1α
2.1. CK1 Family Members
Casein kinases (CKs) were discovered in the 1970s as cytoplasmic protein kinases purified from rat liver, which were able to phosphorylate casein on Ser and Thr residues [11]. Subsequently, multiple CKs were identified and divided into two major groups, CK1 and CK2, based on their biochemical properties [12]. Besides CK1α, the CK1 family of genes encodes CK1β, δ, ε, γ1, γ2, and γ3 [13]. CK1 family members are broadly expressed throughout development and in numerous adult tissues in humans, except CK1β, which is found only in cattle [13]. The primary sequence alignment of CK1 family members highlights a highly conserved Ser/Thr protein kinase domain flanked by distinct amino-terminal (N-term) and carboxyl-terminal (C-term) extensions. Consistent with their homologous protein kinase domain, CK1 family members exhibit similar substrate specificity in vitro. The consensus phosphorylation motif recognized by CK1 was originally identified as a phosphorylated Ser/Thr residue (pSer/Thr) or an acidic group of amino acids upstream of two to four residues, followed by a Ser/Thr phosphor-acceptor [14]. CK1 is also able to phosphorylate substrates at non-consensus sequences [15][16]. For example, CK1α phosphorylates β-catenin at the first serine residue in a novel serine-leucine-serine (SLS) motif upstream of an acidic cluster of six amino acids [16].
2.2. CK1 in Wnt Signaling
Despite their substrate similarity in vitro, the substrates of CK1 family members likely vary in vivo. Multiple CK1 family members (CK1α, δ, ε, γ1) regulate Wnt signaling, and this regulation occurs via the phosphorylation of distinct substrates [17]. CK1δ and CK1ε share the highest primary sequence identity and can play a redundant function, such as the phosphorylation and positive regulation of Dvl [18]. CK1ε and CK1γ1 also play positive roles in Wnt signaling, respectively, by phosphorylating TCF3 to enhance its activity [19] or phosphorylating LRP5/6 to enhance signal transduction [20]. In contrast to other CK1 family members, CK1α plays a negative role in Wnt pathway regulation [21][22]. In addition to its well-established role in the cytosolic β-catenin destruction complex, CK1α also regulates the steady-state levels of nuclear Pygo to attenuate β-catenin/TCF-driven Wnt pathway activity.
2.3. CK1α Splice Variants
The CK1α gene, CSNK1A1, undergoes alternative splicing to produce four splice variants [23][24][25]. These splice variants are distinguished by the absence or presence of a long insert (L) of 28 amino acids in the protein kinase domain or a short insert (S) of 12 amino acids near the C-terminus. In human, the four CK1α splice variants include CK1α with both L and S inserts (CK1αLS), CK1α with only an S insert (CK1αS), CK1α with no insert (CK1αNI), and CK1αLS with a truncated N-term (CK1αSN) (Figure 1
). The L insert contains a nuclear localization signal (NLS), leading to the preferential nuclear enrichment of CK1α splice variants with this insert [25][26]. CK1α splice variants also exhibit other distinct biological properties, including kinetic characteristics, response to small-molecule modulators, thermal stability, and autophosphorylation [23][24][25][27]. In cells, ectopic expression of the various CK1α splice variants leads to varying phosphorylation of cellular β-catenin on Ser45, suggesting that CK1α splice variants might also affect Wnt pathway activity differentially [28].
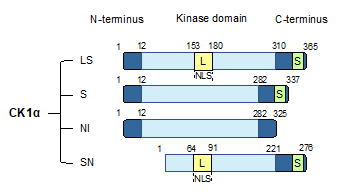
Figure 1. CK1α splice variants. Human CK1α undergoes alternative splicing to produce four splice variants, as shown. These CK1α splice variants are characterized by the insertion of two polypeptide sequences: a long insertion (L) that contains a nuclear localization signal (NLS) into the protein kinase domain, and a short insertion (S) close to the C-terminus. LS: CK1α with both L and S inserts; S: CK1α with only an S insert; NI: CK1α with no insert; SN: CK1α LS with an N-terminal truncation.
2.4. Regulation of CK1α
Although the mechanisms by which CK1α regulates cellular processes, including Wnt signaling, are well established, the regulation of CK1α itself is poorly understood and is thus an active area of investigation. Recently, multiple proteins have been described that regulate the intracellular localization of CK1α (Figure 2) [29][30]. For example, in prostate cancers, glioma pathogenesis-related protein 1 (GLIPR1) mediates the translocation of CK1α to the nucleus, leading to the phosphorylation and degradation of C-Myc and inhibition of Wnt activity [29]. The protein levels of CK1α can also be regulated: family with sequence similarity 83G protein (FAM83G) (also known as protein associated with SMAD1 (PAWS1)) interacts with CK1α in the β-catenin destruction complex and stabilizes CK1α protein, subsequently regulating Wnt signal transduction (Figure 2) [31]. In addition, CK1α gene expression and protein abundance are decreased in many Wnt-driven cancers.
The protein kinase activity of CK1α can be regulated by the presence of DEAD-box RNA helicase 3 (DDX3) in the basal state—upon loss of DDX3, CK1α kinase activity is decreased in cells (Figure 2) [32]. However, whether DDX3 directly regulates CK1α is unclear. The P53 inhibitor protein murine double minute X (MDMX) can also inhibit CK1α’s kinase activity upon their binding, resulting in the activation of Wnt signaling (Figure 2) [33]. This result suggests that MDMX, which binds to CK1α in a stoichiometric fashion, functions as a regulatory subunit for CK1α in Wnt signaling. CK1α is also capable of autophosphorylation, which limits its own kinase activity in vitro. Although the autophosphorylation of CK1δ/ε can be reversed by Wnt signaling, there is currently no evidence showing that CK1α autophosphorylation can be regulated by Wnt signaling [34].
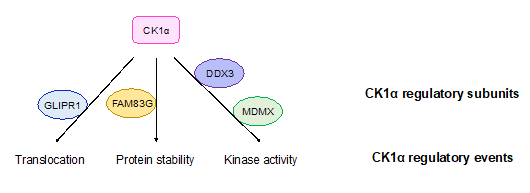
Figure 2. CK1α regulatory subunits. Proteins that have been reported to regulate CK1α are shown. These proteins bind to CK1α and lead to indicated regulatory outcomes of CK1α. GLIPR1: glioma pathogenesis-related protein 1; FAM83G: family with sequence similarity 83G protein; DDX3: DEAD-box RNA helicase 3; MDMX: murine double minute X.
3. CK1α Activators
3.1. Pyrvinium
Based on its important negative role in Wnt signaling, pharmacological activation of CK1α should attenuate Wnt activity. The FDA-approved anthelmintic drug pyrvinium has been the first-in-class small-molecule CK1α activator (Figure 3A), having been identified as a Wnt pathway inhibitor in a large-scale screen of FDA-approved drugs in Xenopus laevis embryo extracts. Importantly, pyrvinium has no observable effect on other pathways examined. CK1α is identified as the target of pyrvinium using a candidate approach and then validated in multiple ways. Although it is shown to bind to multiple CK1 family members, pyrvinium only activates the protein kinase activity of CK1α, consistent with pyrvinium acting as a pharmacological CK1α activator [28]. Pyrvinium, but not its structural analog VU-WS211 (Figure 3A), activates CK1α by increasing its Vmax, without changing its Km for its substrate [28]. These results suggest that pyrvinium activation of CK1α increases the catalytic activity of CK1α without affecting substrate binding, potentially through an allosteric mechanism (Figure 3B) [28]. Interestingly, when comparing the activity of cells transfected with plasmids expressing the four CK1α splice variants, pyrvinium is only able to enhance the activity of those variants lacking the L insert and activated CK1αS the most [28]. Given the location of the L insert within CK1α’s protein kinase domain, which is close to its activation loop (AA 156–190) [35], this result suggests that the L insert may interfere with the binding of pyrvinium to the active site of CK1α.
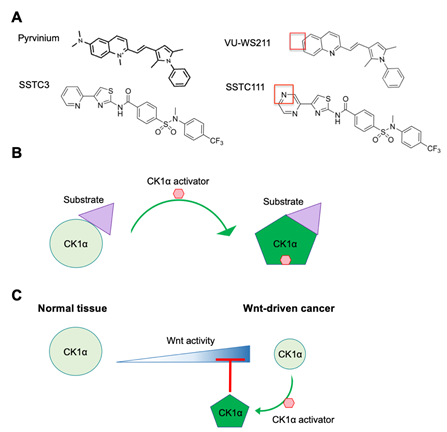
Figure 3. CK1α activators. (A) The structures of two chemically distinct CK1α activators—pyrvinium and SSTC3—and their inactive analogs—VU-WS211 and SSTC111—respectively, are shown. The red boxes highlight key structures needed for maximal efficacy. (B) A model, highlighting how CK1α activators function to increase the catalytic efficiency of CK1α. (C) A model of the mechanism underlying the differential therapeutic index of CK1α activators in normal tissue and Wnt-driven cancer.
Consistent with the pivotal role of CK1α in Wnt signaling, pyrvinium attenuates the growth of Wnt-dependent CRC cell lines in a CK1α-dependent manner. Pyrvinium also exhibits efficacy against a number of other Wnt-driven cancer cell lines, including those derived from breast cancer and hepatocellular carcinoma [36]. Pyrvinium inhibits Wnt pathway activity by reducing β-catenin levels in the cytoplasm and by increasing the degradation of the β-catenin/TCF coactivator Pygo in the nucleus. Consistent with Pygo being a relevant nuclear target of CK1α in Wnt signaling, pyrvinium is able to attenuate the growth of a CRC cell line, harboring a constitutively active β-catenin oncogenic mutant that lacks the CK1α phospho-acceptor Ser. Despite its potent Wnt-inhibiting effect ex vivo, the subsequent evaluation of pyrvinium’s efficacy against the growth of Wnt-driven cancers in vivo has been limited, as pyrvinium has low bioavailability outside of the intestinal tract[37]. However, pyrvinium has been evaluated in a Wnt-dependent FAP-induced colorectal adenoma mouse model, in which it significantly decreases the formation of adenomatous polyps in an on-target manner. Based on this work, pyrvinium has been designated by the FDA as an orphan drug for the treatment of FAP.
3.2. SSTC Compounds
A number of potent, chemically novel CK1α activators (SSTC3 and SSTC104- see Figure 3), which have significantly improved bioavailability by comparison with pyrvinium, have been described. These second-generation CK1α activators bind to CK1α in a manner that is competitive to pyrvinium, suggesting that they bind to a similar site on CK1α . They also attenuate Wnt activity in a manner that is dependent on CK1α. SSTC3, but not its structural analog SSTC111 (Figure 3), inhibits the growth of Wnt-driven CRC cell lines and patient-derived CRC organoids ex vivo . Consistent with its improved bioavailability, SSTC3 remains in plasma for 24 h after intraperitoneal injection in mice and is able to penetrate the blood-brain barrier [38]. Furthermore, SSTC3 significantly inhibits the growth of primary and metastatic tumors that develop from patient CRCs or a Wnt-driven CRC cell line implanted in mice. Importantly, when used at a dose that is efficacious against CRC growth, SSTC3 exhibits no significant toxicity in mice. Specifically, the structure of normal intestinal tissue, which is one of the sites primarily impeded from on-target toxicity of most Wnt inhibitors, is not disrupted by SSTC3 treatment. It has been proposed that the greater therapeutic index of SSTC3 compared to other Wnt inhibitors is the result of the decreased abundance of CK1α protein in CRC tissue versus normal intestinal tissue (Figure 3C). Thus, these reduced levels of CK1α protein sensitize CRC cells to the enhanced activation of CK1α in response to SSTC3.
References
- Clevers, H. Wnt/beta-catenin signaling in development and disease. Cell 2006, 127, 469–480, doi:10.1016/j.cell.2006.10.018.
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, doi:10.1242/dev.146589.
- Nusse, R.; Brown, A.; Papkoff, J.; Scambler, P.; Shackleford, G.; McMahon, A.; Moon, R.; Varmus, H. A new nomenclature for int-1 and related genes: The Wnt gene family. Cell 1991, 64, 231, doi:10.1016/0092-8674(91)90633-a.
- Mikels, A.J.; Nusse, R. Wnts as ligands: Processing, secretion and reception. Oncogene 2006, 25, 7461–7468, doi:10.1038/sj.onc.1210053.
- Willert, K.; Nusse, R. Wnt proteins. Cold Spring Harb. Perspect. Biol. 2012, 4, a007864, doi:10.1101/cshperspect.a007864.
- Zhai, L.; Chaturvedi, D.; Cumberledge, S. Drosophila wnt-1 undergoes a hydrophobic modification and is targeted to lipid rafts, a process that requires porcupine. J. Biol. Chem. 2004, 279, 33220–33227, doi:10.1074/jbc.M403407200.
- van den Heuvel, M.; Harryman-Samos, C.; Klingensmith, J.; Perrimon, N.; Nusse, R. Mutations in the segment polarity genes wingless and porcupine impair secretion of the wingless protein. EMBO J. 1993, 12, 5293–5302.
- Kadowaki, T.; Wilder, E.; Klingensmith, J.; Zachary, K.; Perrimon, N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 1996, 10, 3116–3128, doi:10.1101/gad.10.24.3116.
- Hofmann, K. A superfamily of membrane-bound O-acyltransferases with implications for wnt signaling. Trends Biochem. Sci. 2000, 25, 111–112, doi:10.1016/s0968-0004(99)01539-x.
- Willert, K.; Brown, J.D.; Danenberg, E.; Duncan, A.W.; Weissman, I.L.; Reya, T.; Yates, J.R., 3rd; Nusse, R. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003, 423, 448–452, doi:10.1038/nature01611.
- Matsumura, S.; Takeda, M. Phosphoprotein kinases from rat liver cytosol. Biochim. Biophys. Acta 1972, 289, 237–241, doi:10.1016/0005-2744(72)90127-1.
- Hathaway, G.M.; Traugh, J.A. Cyclic nucleotide-independent protein kinases from rabbit reticulocytes. Purification of casein kinases. J. Biol. Chem. 1979, 254, 762–768.
- Knippschild, U.; Gocht, A.; Wolff, S.; Huber, N.; Lohler, J.; Stoter, M. The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell Signal. 2005, 17, 675–689, doi:10.1016/j.cellsig.2004.12.011.
- Flotow, H.; Graves, P.R.; Wang, A.Q.; Fiol, C.J.; Roeske, R.W.; Roach, P.J. Phosphate groups as substrate determinants for casein kinase I action. J. Biol. Chem. 1990, 265, 14264–14269.
- Xu, Y.; Lee, S.H.; Kim, H.S.; Kim, N.H.; Piao, S.; Park, S.H.; Jung, Y.S.; Yook, J.I.; Park, B.J.; Ha, N.C. Role of CK1 in GSK3beta-mediated phosphorylation and degradation of snail. Oncogene 2010, 29, 3124–3133, doi:10.1038/onc.2010.77.
- Marin, O.; Bustos, V.H.; Cesaro, L.; Meggio, F.; Pagano, M.A.; Antonelli, M.; Allende, C.C.; Pinna, L.A.; Allende, J.E. A noncanonical sequence phosphorylated by casein kinase 1 in beta-catenin may play a role in casein kinase 1 targeting of important signaling proteins. Proc. Natl. Acad. Sci. USA. 2003, 100, 10193–10200, doi:10.1073/pnas.1733909100.
- Cruciat, C.M. Casein kinase 1 and Wnt/beta-catenin signaling. Curr. Opin. Cell. Biol. 2014, 31, 46–55, doi:10.1016/j.ceb.2014.08.003.
- Peters, J.M.; McKay, R.M.; McKay, J.P.; Graff, J.M. Casein kinase I transduces Wnt signals. Nature 1999, 401, 345–350, doi:10.1038/43830.
- Lee, E.; Salic, A.; Kirschner, M.W. Physiological regulation of [beta]-catenin stability by Tcf3 and CK1epsilon. J. Cell Biol. 2001, 154, 983–993, doi:10.1083/jcb.200102074.
- Davidson, G.; Wu, W.; Shen, J.; Bilic, J.; Fenger, U.; Stannek, P.; Glinka, A.; Niehrs, C. Casein kinase 1 gamma couples Wnt receptor activation to cytoplasmic signal transduction. Nature 2005, 438, 867–872, doi:10.1038/nature04170.
- Lebensohn, A.M.; Dubey, R.; Neitzel, L.R.; Tacchelly-Benites, O.; Yang, E.; Marceau, C.D.; Davis, E.M.; Patel, B.B.; Bahrami-Nejad, Z.; Travaglini, K.J.; et al. Comparative genetic screens in human cells reveal new regulatory mechanisms in WNT signaling. Elife 2016, 5, doi:10.7554/eLife.21459.
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. beta-Catenin-driven cancers require a YAP1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473, doi:10.1016/j.cell.2012.11.026.
- Yong, T.J.; Gan, Y.Y.; Toh, B.H.; Sentry, J.W. Human CKIalpha(L) and CKIalpha(S) are encoded by both 2.4- and 4. 2-kb transcripts, the longer containing multiple RNA-destablising elements. Biochim. Biophys. Acta 2000, 1492, 425–433, doi:10.1016/s0167-4781(00)00146-9.
- Zhang, J.; Gross, S.D.; Schroeder, M.D.; Anderson, R.A. Casein kinase I alpha and alpha L: Alternative splicing-generated kinases exhibit different catalytic properties. Biochemistry 1996, 35, 16319–16327, doi:10.1021/bi9614444.
- Burzio, V.; Antonelli, M.; Allende, C.C.; Allende, J.E. Biochemical and cellular characteristics of the four splice variants of protein kinase CK1alpha from zebrafish (Danio rerio). J. Cell Biochem. 2002, 86, 805–814, doi:10.1002/jcb.10263.
- Fu, Z.; Chakraborti, T.; Morse, S.; Bennett, G.S.; Shaw, G. Four casein kinase I isoforms are differentially partitioned between nucleus and cytoplasm. Exp. Cell Res. 2001, 269, 275–286, doi:10.1006/excr.2001.5324.
- Budini, M.; Jacob, G.; Jedlicki, A.; Perez, C.; Allende, C.C.; Allende, J.E. Autophosphorylation of carboxy-terminal residues inhibits the activity of protein kinase CK1alpha. J. Cell Biochem. 2009, 106, 399–408, doi:10.1002/jcb.22019.
- Shen, C.; Li, B.; Astudillo, L.; Deutscher, M.P.; Cobb, M.H.; Capobianco, A.J.; Lee, E.; Robbins, D.J. The CK1alpha activator pyrvinium enhances the catalytic efficiency (kcat/Km) of CK1alpha. Biochemistry 2019, 58, 5102–5106, doi:10.1021/acs.biochem.9b00891.
- Li, L.; Ren, C.; Yang, G.; Fattah, E.A.; Goltsov, A.A.; Kim, S.M.; Lee, J.S.; Park, S.; Demayo, F.J.; Ittmann, M.M.; et al. GLIPR1 suppresses prostate cancer development through targeted oncoprotein destruction. Cancer Res. 2011, 71, 7694–7704, doi:10.1158/0008-5472.CAN-11-1714.
- Fulcher, L.J.; Bozatzi, P.; Tachie-Menson, T.; Wu, K.Z.L.; Cummins, T.D.; Bufton, J.C.; Pinkas, D.M.; Dunbar, K.; Shrestha, S.; Wood, N.T.; et al. The DUF1669 domain of FAM83 family proteins anchor casein kinase 1 isoforms. Sci. Signal. 2018, 11, doi:10.1126/scisignal.aao2341.
- Bozatzi, P.; Dingwell, K.S.; Wu, K.Z.; Cooper, F.; Cummins, T.D.; Hutchinson, L.D.; Vogt, J.; Wood, N.T.; Macartney, T.J.; Varghese, J.; et al. PAWS1 controls Wnt signalling through association with casein kinase 1alpha. EMBO Rep. 2018, 19, doi:10.15252/embr.201744807.
- Cruciat, C.M.; Dolde, C.; de Groot, R.E.; Ohkawara, B.; Reinhard, C.; Korswagen, H.C.; Niehrs, C. RNA helicase DDX3 is a regulatory subunit of casein kinase 1 in Wnt-beta-catenin signaling. Science 2013, 339, 1436–1441, doi:10.1126/science.1231499.
- Huang, Q.; Chen, L.; Schonbrunn, E.; Chen, J. MDMX inhibits casein kinase 1alpha activity and stimulates Wnt signaling. EMBO J. 2020, 10.15252/embj.2020104410, e104410, doi:10.15252/embj.2020104410.
- Swiatek, W.; Tsai, I.C.; Klimowski, L.; Pepler, A.; Barnette, J.; Yost, H.J.; Virshup, D.M. Regulation of casein kinase I epsilon activity by Wnt signaling. J. Biol. Chem. 2004, 279, 13011–13017, doi:10.1074/jbc.M304682200.
- Xu, R.M.; Carmel, G.; Sweet, R.M.; Kuret, J.; Cheng, X. Crystal structure of casein kinase-1, a phosphate-directed protein kinase. EMBO J. 1995, 14, 1015–1023.
- Barham, W.; Frump, A.L.; Sherrill, T.P.; Garcia, C.B.; Saito-Diaz, K.; VanSaun, M.N.; Fingleton, B.; Gleaves, L.; Orton, D.; Capecchi, M.R.; et al. Targeting the Wnt pathway in synovial sarcoma models. Cancer Discov. 2013, 3, 1286–1301, doi:10.1158/2159-8290.CD-13-0138.
- Smith, T.C.; Kinkel, A.W.; Gryczko, C.M.; Goulet, J.R. Absorption of pyrvinium pamoate. Clin. Pharmacol Ther. 1976, 19, 802–806, doi:10.1002/cpt1976196802.
- Rodriguez-Blanco, J.; Li, B.; Long, J.; Shen, C.; Yang, F.; Orton, D.; Collins, S.; Kasahara, N.; Ayad, N.G.; McCrea, H.J.; et al. A CK1alpha activator penetrates the brain and shows efficacy against drug-resistant metastatic medulloblastoma. Clin. Cancer Res. 2019, 25, 1379–1388, doi:10.1158/1078-0432.CCR-18-1319.

