Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ALESSANDRO REGGIANI | + 3035 word(s) | 3035 | 2022-01-07 02:50:51 | | | |
| 2 | Bruce Ren | Meta information modification | 3035 | 2022-01-17 02:03:56 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Reggiani, A. A Functional Minigenome of Parvovirus B19. Encyclopedia. Available online: https://encyclopedia.pub/entry/18247 (accessed on 25 June 2026).
Reggiani A. A Functional Minigenome of Parvovirus B19. Encyclopedia. Available at: https://encyclopedia.pub/entry/18247. Accessed June 25, 2026.
Reggiani, Alessandro. "A Functional Minigenome of Parvovirus B19" Encyclopedia, https://encyclopedia.pub/entry/18247 (accessed June 25, 2026).
Reggiani, A. (2022, January 14). A Functional Minigenome of Parvovirus B19. In Encyclopedia. https://encyclopedia.pub/entry/18247
Reggiani, Alessandro. "A Functional Minigenome of Parvovirus B19." Encyclopedia. Web. 14 January, 2022.
Copy Citation
Parvovirus B19 (B19V) is a human pathogenic virus of clinical relevance, characterized by a selective tropism for erythroid progenitor cells in bone marrow. Relevant information on viral characteristics and lifecycle can be obtained from experiments involving engineered genetic systems in appropriate in vitro cellular models. Previously, a B19V genome of defined consensus sequence was designed, synthesized and cloned in a complete and functional form, able to replicate and produce infectious viral particles in a producer/amplifier cell system.
parvovirus B19
synthetic genome
genetic engineering
replicon unit
functional complementation
1. Introduction
Within the family Parvoviridae [1], Parvovirus B19 (B19V) is a human pathogenic virus of clinical relevance, responsible for transient or persistent erythroid aplasia, infectious erythema, arthropathies, myocarditis and intrauterine infections, among others [2][3]. The variability in the pathogenetic processes and the resulting clinical outcomes of diverse nature and severity depend on a complex interplay between the viral properties, the characteristics of target cells in the different tissues, and the physiological status and immune response of infected individuals. B19V has a marked tropism for erythroid progenitor cells (EPCs) in the bone marrow, both susceptible and permissive depending on their differentiation and proliferation state [4]. In EPCs, infection normally induces cell cycle arrest and apoptosis [5], thus causing a temporary block in erythropoiesis which can become clinically relevant [6]. Different non-erythroid cell types, including endothelial, stromal, or synovial cells, are also susceptible but mainly non-permissive. In these cells, infection can trigger inflammatory responses and consequent tissue damage [7], and generally results in long-term persistence of viral DNA within tissues [8].
Investigation of viral genetics is fundamental to better understanding of the B19V replication cycle, the virus–cell interaction in different environments, the pathogenetic processes underlying the wide range of associated diseases, and the devising of more efficient antiviral strategies [9][10]. The B19V genome is composed of two inverted, terminal repeated regions (ITR) of imperfect palindromic sequence, 383 nt long, flanking a unique internal region (IR), 4830 nt long, containing all the coding sequences (Figure S1). The role of ITRs is crucial. The ability of palindromic sequences to fold in self-priming, hairpin secondary structures, and the presence of specific cis-recognition sequences acting as origins of replication, allow replication of the viral DNA through a rolling hairpin mechanism [11][12][13]. The activity of the unique transcription promoter (P6) at the left end of the internal region also depends critically on regulatory sequences within the upstream ITR [14][15][16][17][18]. Within the IR, distribution of splicing and cleavage-polyadenylation recognition sequences along the genome ensures coordinate processing of the pre-mRNA to a set of mature mRNAs [19][20][21][22]. Functionally, these can be divided into a set expressed from the left side of the genome, mainly coding for NS1 protein in an early phase of replication, and a set expressed from the right side of the genome, mainly coding for the structural VP proteins in the late phase of replication [23][24][25].
NS1 protein is the major non-structural protein, essential to virus replication and central for interaction with host cell components [9]. It is involved in the replication of the B19V genome, by its capacity to bind to specific recognition sites in the terminal regions, and by its endonuclease and helicase activities effecting ITR terminal resolution and strand unwinding. It is involved in viral transcription, enhancing activity of the P6 promoter by its trans-activating domains, thus promoting overall B19V genome expression. Besides, NS1 is a heterologous trans-activator of cellular genes, therefore inducing alterations in the cellular environment. It has a role in regulating progression through the cell cycle and in inducing apoptosis, therefore contributing to the pathogenesis of B19V infection. Given all this, NS1 is a matter of relevant interest in studying B19V, not least as a pharmacological target [26][27]. Viral capsid proteins VP1 and VP2 assemble to form a capsid shell of 22 nm in diameter, arranged in a T = 1 icosahedral structure [28]. Being translated from the same coding frame, both proteins share a common region that forms the core shell, while the VP1 protein, about 5% in abundance, possesses an additional N-terminal region, VP1u, crucial for cell recognition, attachment and penetration [29][30].
Information can be obtained from experiments involving engineered genetic systems in appropriate in vitro cellular models, since cloned forms of the B19V genome can be competent for replication and constitute an effective tool for studying the viral lifecycle and its interaction with target cells [31][32]. Previously, a model system was established by a novel synthetic strategy [33]. A reference genome of defined consensus sequence was designed, synthesized and cloned in a complete and functional form in a plasmid vector. Such a genome was able to replicate and produce infectious viral particles in a producer/amplifier cell system, the myeloblastoid UT7/EpoS1cells, allowing generation and further propagation of virus in EPC cell cultures. Replicative competence was linked to preservation of sequence integrity and asymmetry within the terminal regions, while preservation of the complete internal region ensured maintenance of the cis-acting signals required for regulation of viral genome expression. Therefore, the full proteome of B19V could be co-ordinately expressed and novel infectious viral particles produced. Further investigation is required to assess the flexibility of this system to genetic manipulation, and its potential as an advanced tool for basic research and translational applications.
2. Design and Construction of a B19V Minigenome
In the design of a B19V minigenome with potential replicative activity, the rational requirements were: (i) to preserve both terminal regions up to the sites of dyad symmetry, retaining the capacity of hairpin formation; and (ii) to preserve the internal region extending up to the pAp1 proximal cleavage-polyadenylation signal, as a gene cassette with potential for coding for the NS1 protein, while eliminating the genomic region coding for the viral capsid proteins. To this end, a large deletion was operated in the right-side of genome, and a novel chimeric cleavage-polyadenylation signal created, named pAs1, joining the upstream cis-elements of pAp1 and the downstream cis-elements of pAd (Figure 1).
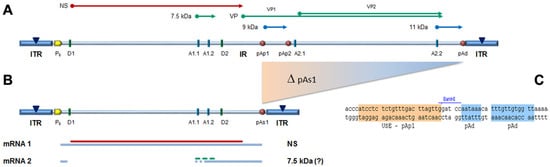
Figure 1. (A) Map of B19V genome. ITR: inverted terminal repeats (▼, site of dyad symmetry). IR: internal region and relevant cis-acting functional sites (P6, promoter; pAp1, pAp2, proximal cleavage-polyadenylation sites; pAd, distal cleavage-polyadenylation site; D1, D2, splice donor sites; A1.1, A1.2, A2.1, A2.2, splice acceptor sites). Coding sequences for viral NS, VP and smaller non-structural proteins are aligned to map. Δ: deletion to create a novel cleavage-polyadenylation signal (pAs1). (B) Map of B19V derived minigenome; simplified transcription map, indicating the two classes of mRNAs (mRNA 1–2), with alternative splicing forms (dashed lines) and related coding potential. (C) Sequence at the novel pAs1 cleavage-polyadenylation site (USE—pAp1, upstream element to pAp1 [19]).
For the construction of the minigenome, a synthetic gene segment encompassing the designed deletion substituted by the novel pAs1 sequence was synthesised and inserted in order to replace the original sequence in the previously established pCK10 and pCH10 plasmids [33]. The viral insert in pCK10 preserves both terminal regions extending beyond the site of dyad symmetry; however, attempts at cloning in pCK10 only yielded unstable plasmid clones, with deletion of the palindromic sequence in the right-hand terminal region. The viral insert in pCH10 preserves both terminal regions extending up to the site of dyad symmetry; in this case, a stable plasmid clone was successfully obtained, named pCH10-pAs1.
3. Functional Competence of the B19V Minigenome
From the pCH10 plasmid, three genomic inserts of different extension can be obtained, differing in their functional competence: CH10, corresponding to the whole cloned insert, extending in the terminal regions to the sites of dyad symmetry (nt. 184–5413); CI0, extending in the terminal regions within the sites of dyad symmetry (nt. 245–5474); and CJ0, extending in the terminal regions only to the start of palindromes (nt. 366–5231). From the pCH10-pAs1 plasmid, three genomic inserts of corresponding extension could also be obtained by analogy: CH10-pAs1, CI0-pAs1, and CJ0-pAs1. The biological activity and functional competence of pCH10 and pCH10-pAs1 derived inserts was comparatively analysed following transfection in UT7/EpoS1 cells. Genomic inserts were obtained by means of in vitro amplification, then purified inserts were used to transfect UT7/EpoS1 cells. At 8- and 24-h post-transfection (hpt), aliquots of cell culture were sampled for quantification of viral nucleic acids (DNA, mRNAs) by qPCR and qRT-PCR (Figure 2), and detection of the NS protein by IIF and cytofluorimetric analysis.
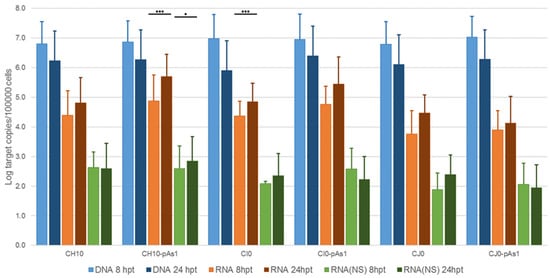
Figure 2. Viral nucleic acids in UT7/EpoS1 cells, transfected with CH10 and CH10-pAs1 derived inserts. Log amounts of target copies (viral DNA, total RNA, NS1 mRNA), normalized to 105 cells, at 8 and 24 hpt. Mean and std of duplicate determinations for two different experiments. Two-way ANOVA, Bonferroni post-test: ***, p < 0.001; *, p < 0.05.
The amount of DNA, either at 8 or 24 hpt, was comparable for all tested inserts, indicating a similar transfection efficiency. Due to the large quantity of input DNA used in transfection, no significant temporal variation in DNA amount was observed for any of the tested inserts, apart from a general decrease from 8 to 24 hpt (mean 0.70 Log, range −1.1–0.57), likely to be due to progressive degradation of exogenous DNA. De novo synthesis of transfected DNA was assessed in a parallel experiment, by transfection of inserts directly excised from plasmids and tested for Dam methylation pattern and resistance to DpnI cleavage, both by a qPCR assay and a Southern Blot analysis. In this way, it was possible to investigate CH10 and CH10-pAs1, excised using BssHII, CI0 and CI0-pAs1, excised using AccIII, but not CJ0 and CJ0-pAs1, because of lack of corresponding RE sites.
By qPCR, DNA excised from plasmids was resistant to 1.7% and 1.4% for CH10 and CH10-pAs1, and 1.6% and 1.3% for CI0 and CI0-pAs1. DNA obtained from transfected cells at 24 hpt was resistant to 11.7% and 15.8%, and 8.8% and 19.0%, respectively. By Southern Blot (Figure 3), at 24 hpt, bands corresponding to full-length, DpnI resistant DNA were also observed for all transfected inserts. Data thus obtained are consistent with the maintenance of the replicative competence of transfected inserts, at least for CH10-and CI0-derived inserts, both the complete genomes and the derived minigenomes, a property likely to be due to the preservation of sequence symmetry within the terminal regions and implying a hairpin-independent priming of DNA synthesis.
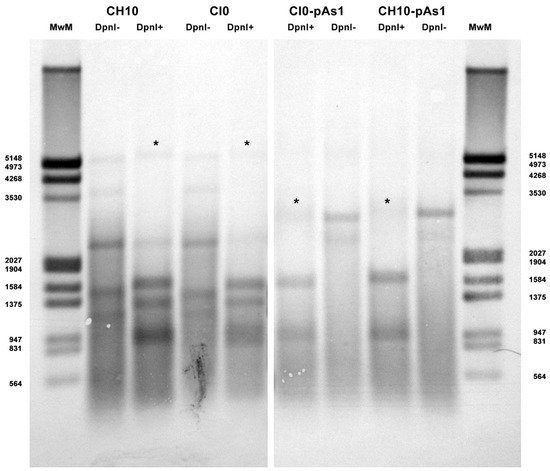
Figure 3. Southern Blot Analysis of B19V DNA obtained from UT7/EpoS1 cells transfected with inserts CH10, CI0 and CH10-pAs1, CI0-pAs1, collected at 24 hpt. Samples were treated by RE DpnI to distinguish de novo synthesized viral DNA (*) based on different dam methylation pattern and sensitivity to RE DpnI. MwM: molecular weight marker III, Dig-labelled (Roche). Southern Blotting and hybridization using a full-length digoxigenin-labelled DNA probe was carried out as described [33].
All transfected inserts showed a sustained transcriptional activity. Viral mRNAs were detected for all inserts at both time-points post-transfection, with a general increase from 8 to 24 hpt of total mRNA (mean 0.56 Log, range −0.01–1.33), and to a lesser extent of NS mRNA (mean 0.08 Log, range −0.36–0.51), significant for CH10-pAs1 only. Although with some variability, overall results attested the early onset and maintenance of viral transcription, implying processing of pre-mRNA at the novel chimeric pAs1 cleavage-polyadenylation site, and preservation of a balanced usage of splicing signals. In fact, a typical ratio of mRNAs pertaining to the left-side genome was produced, including both the unspliced mRNAs coding for NS protein (mRNA 1 in Figure 1), approximately 1% of total viral mRNAs, and the more abundant spliced mRNAs (mRNA 2 in Figure 1), in a pattern analogous to that observed in the early phase of the replicative cycle.
The expression of NS protein was monitored by IIF and cytofluorimetric analysis, sampling transfected cell cultures at 8 and 24 hpt. By IIF (Figure 4), NS protein was already observed at 8 hpt, and at 24 hpt the number of positive cells and signal intensity both increased. Distribution of the protein within the cells showed the same nuclear/cytoplasmic pattern both for all complete or derived minigenome inserts.
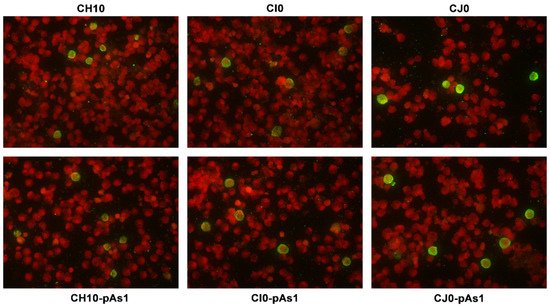
Figure 4. UT7/EpoS1 cells transfected with the indicated inserts were sampled at 24 hpt, and NS1 protein was detected by IIF. Original magnification 400×.
By cytofluorimetric analysis (Figure 5), a quantitative assessment of cells expressing NS1 protein was obtained at 24 hpt. The percentage of positive cells was in the range 0.2–1.3% for the complete inserts and increased to 2.6–5.0% for the derived minigenomes, the highest increase was observed for the CI0/CI0-pAs1 combination. Altogether, experiments suggest that the lower genetic complexity of modified genomes promoted progressive expression and accumulation of NS protein in an increasing fraction of the cell population.
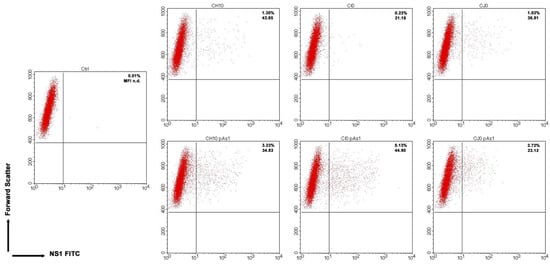
Figure 5. UT7/EpoS1 cells transfected with the indicated inserts (Ctrl, no DNA control) were sampled at 24 hpt, and cell population was analysed by cytofluorimeter to determine the percentage of NS1 expressing cells. Dot plot graph on gated cell population for FSc and NS1 FITC. Reported percentage values of positive cells and geometric mean fluorescence intensity (MFI) for positive subpopulations reported as the average result of two independent determinations.
4. Functional Complementation of the B19V Minigenome
The capacity of the CH10-pAs1 minigenome to provide complementing functions through expression of NS1 protein was thereafter tested in a subsequent series of experiments. To the purpose, a set of modified B19V genome clones was designed, defective for the coding sequence of NS protein. In particular, the pCH10 clone was modified by deletion, removing the genomic region corresponding to the first intron within the NS gene, from the splice donor site D1 to the two possible alternative splice acceptor sites A1.1 and A1.2. The obtained plasmids, named CH10-A1.1 and CH10-A1.2, retained the cis-acting sequences directing alternative cleavage-polyadenylation at pAp and pAd sites, sequences regulating alternative splicing of the distal introns at D2 and A2.1/2 sites, and all of the coding sequences for the VP1, VP2 and 11 kDa proteins (Figure 6).
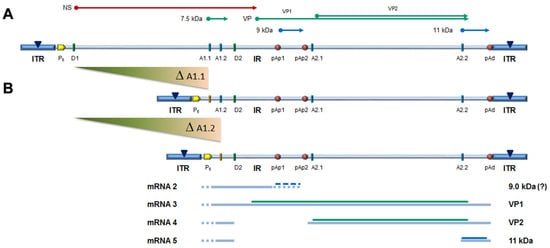
Figure 6. (A) Map of B19V genome, see Figure 1. Δ: deletion to remove first intron (A1.1/2). (B) Map of B19V derived minigenomes; simplified transcription map, indicating the four classes of mRNAs (mRNA 2–5), with alternative splicing/cleavage forms (dashed lines) and related coding potential.
To evaluate the functional competence and possible complementation effects for these defective genomes, inserts obtained by means of in vitro amplification were transfected in UT7/EpoS1 cells, alone or in co-transfection with CH10-pAs1 as helper plasmid. At 24 hpt, aliquots of cell culture were sampled for quantification of viral nucleic acids (DNA, mRNAs) by qPCR and qRT-PCR (Figure 7), and detection of the NS1 and VP proteins by IIF.
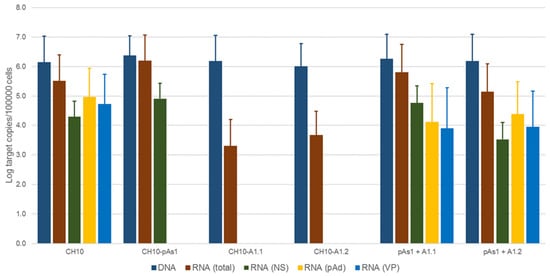
Figure 7. Viral nucleic acids in UT7/EpoS1 cells, transfected/co-transfected with CH10, CH10-pAs1 and CH10-A1.1/2 inserts. Log amounts of target copies (viral DNA, total RNA, NS1 mRNA, pAd cleaved RNA, VP RNA), normalized to 105 cells, at 24 hpt. Mean and std of duplicate determinations for two different experiments.
No significant differences were observed in the amounts of viral DNA, but relevant information was obtained by quantification of viral RNA. By comparison to the reference CH10 insert, insert CH10-pAs1 confirmed its high transcriptional activity and correct mRNA processing, with an abundance of about 1% unspliced mRNAs coding for NS protein. The NS defective, CH10-A1.1 and -A.2 inserts showed a reduced (−3 Log) basal transcriptional activity, which allowed detection of only the proximally cleaved mRNAs (mRNA 2 in Figure 6), and not of any distally cleaved mRNA (mRNAs 3–5 in Figure 5). Cotransfection of these inserts with CH10-pAs1 inset led to functional complementation, restoring expression from the NS-defective genomes, as shown by the detection of A1.1 and A.2 derived mRNAs, to amounts only about 1 Log lower than what observed for the reference CH10 insert. By composition, the most abundant mRNAs were still the proximally cleaved mRNA species (mRNA 2), which were contributed by both CH10-pAs1 (following splicing) and A1.1/2 (following cleavage at pAp); abundance of NS mRNA (mRNA 1), contributed only by CH10-pAs1, pAd cleaved mRNAs (mRNA 3–5) and VP mRNAs (mRNA 3–4), contributed only by A1.1/2, were in all cases in the order of 1% of total mRNAs.
By IIF analysis (Figure 8), expression of both NS and to lesser extents VP proteins was confirmed for the CH10 insert. The expression of NS protein was also confirmed for CH10-pAs1, and not detected in the case of CH10-A1.1/2, as expected. Expression of VP proteins from CH10-A1.1/2 inserts alone was not observed. Cotransfection of CH10-pAs1 and A1.1/2 preserved expression of NS protein from CH10-pAs1 and restored the expression of VP proteins from CH10-A1.1/2, although for the latter detection was limited to a small number of cells. Altogether, data confirm that the NS protein produced by the CH10-pAs1 minigenome is functional in complementation of defective B19V genomes, restoring expression of the late set of mRNAs to a pattern similar to the standard expression profile of B19V genome and allowing production of capsid proteins.
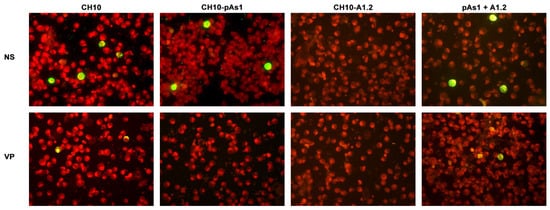
Figure 8. UT7/EpoS1 cells transfected with the indicated inserts were sampled at 24 hpt, and NS1 or VP1/2 proteins were detected by IIF. Results obtained for insert CH10-A1.1 were analogous to what obtained for CH10-A1.2 (not in figure). Original magnification 400×.
5. Extracellular Vehiculation of Minigenomes
Following transfection of UT7/EpoS1 cells, measurable amounts of viral DNA were detectable in the cell culture medium until 6 days post-transfection, not associated to cells but partially resistant to nuclease treatment. The possibility that this genetic material could be transferrable to susceptible EPCs was investigated. For the purpose, CH10-pAs1 and CH10-A1.1/2 inserts were transfected in UT7/EpoS1 cells in the different combinations as described, and expression of NS and VP proteins first confirmed for all competent combinations. Then, after a 6 day course, the supernatant of transfected cell cultures was collected and added to in vitro differentiated EPCs cells, as a test system. After a further 48 h course of incubation, EPCs were collected and analysed for any presence of B19V DNA, RNA or expression of NS protein. For all experimental samples, a low measurable amount of DNA was found associated to EPCs (<102 copies/105 cells). However, no transcriptional activity could be detected in EPCs in any tested combination and no NS protein production could be observed. The obtained results imply that the vehiculation of genetic material from cells transfected with the CH10 derived clones may not be due to the formation of transducing viral particles, and that the process is not functionally relevant to a measurable extent. The hypothesis that such transfer should be attributed to simple carry-over in a nuclease-resistance form, or to the formation of extracellular vesicles or exosomes, and whether a limited number of transducing viral particles is actually produced, requires further investigation.
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Penzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368.
- Gallinella, G. Parvovirus B19 Achievements and Challenges. ISRN Virol. 2013, 2013, 898730.
- Qiu, J.; Soderlund-Venermo, M.; Young, N.S. Human Parvoviruses. Clin. Microbiol. Rev. 2017, 30, 43–113.
- Chisaka, H.; Morita, E.; Yaegashi, N.; Sugamura, K. Parvovirus B19 and the pathogenesis of anaemia. Rev. Med. Virol. 2003, 13, 347–359.
- Chen, A.Y.; Qiu, J. Parvovirus infection-induced cell death and cell cycle arrest. Future Virol. 2010, 5, 731–743.
- Kerr, J.R. A review of blood diseases and cytopenias associated with human parvovirus B19 infection. Rev. Med. Virol. 2015, 25, 224–240.
- Adamson-Small, L.A.; Ignatovich, I.V.; Laemmerhirt, M.G.; Hobbs, J.A. Persistent parvovirus B19 infection in non-erythroid tissues: Possible role in the inflammatory and disease process. Virus Res. 2014, 190, 8–16.
- Bua, G.; Gallinella, G. How does parvovirus B19 DNA achieve lifelong persistence in human cells? Future Virol. 2017, 12, 549–553.
- Ganaie, S.S.; Qiu, J. Recent Advances in Replication and Infection of Human Parvovirus B19. Front. Cell. Infect. Microbiol. 2018, 8, 166.
- Manaresi, E.; Gallinella, G. Advances in the Development of Antiviral Strategies against Parvovirus B19. Viruses 2019, 11, 659.
- Guan, W.; Wong, S.; Zhi, N.; Qiu, J. The genome of human parvovirus B19 can replicate in nonpermissive cells with the help of adenovirus genes and produces infectious virus. J. Virol. 2009, 83, 9541–9553.
- Luo, Y.; Qiu, J. Human parvovirus B19: A mechanistic overview of infection and DNA replication. Future Virol. 2015, 10, 155–167.
- Zou, W.; Wang, Z.; Xiong, M.; Chen, A.Y.; Xu, P.; Ganaie, S.S.; Badawi, Y.; Kleiboeker, S.; Nishimune, H.; Ye, S.Q.; et al. Human Parvovirus B19 Utilizes Cellular DNA Replication Machinery for Viral DNA Replication. J. Virol. 2018, 92, e01881-17.
- Gareus, R.; Gigler, A.; Hemauer, A.; Leruez-Ville, M.; Morinet, F.; Wolf, H.; Modrow, S. Characterization of cis-acting and NS1 protein-responsive elements in the p6 promoter of parvovirus B19. J. Virol. 1998, 72, 609–616.
- Raab, U.; Bauer, B.; Gigler, A.; Beckenlehner, K.; Wolf, H.; Modrow, S. Cellular transcription factors that interact with p6 promoter elements of parvovirus B19. J. Gen. Virol. 2001, 82, 1473–1480.
- Raab, U.; Beckenlehner, K.; Lowin, T.; Niller, H.H.; Doyle, S.; Modrow, S. NS1 protein of parvovirus B19 interacts directly with DNA sequences of the p6 promoter and with the cellular transcription factors Sp1/Sp3. Virology 2002, 293, 86–93.
- Bonvicini, F.; Manaresi, E.; Di Furio, F.; De Falco, L.; Gallinella, G. Parvovirus B19 DNA CpG dinucleotide methylation and epigenetic regulation of viral expression. PLoS ONE 2012, 7, e33316.
- Bua, G.; Tedesco, D.; Conti, I.; Reggiani, A.; Bartolini, M.; Gallinella, G. No G-Quadruplex Structures in the DNA of Parvovirus B19: Experimental Evidence versus Bioinformatic Predictions. Viruses 2020, 12, 935.
- Yoto, Y.; Qiu, J.; Pintel, D.J. Identification and characterization of two internal cleavage and polyadenylation sites of parvovirus B19 RNA. J. Virol. 2006, 80, 1604–1609.
- Guan, W.; Cheng, F.; Yoto, Y.; Kleiboeker, S.; Wong, S.; Zhi, N.; Pintel, D.J.; Qiu, J. Block to the production of full-length B19 virus transcripts by internal polyadenylation is overcome by replication of the viral genome. J. Virol. 2008, 82, 9951–9963.
- Guan, W.; Huang, Q.; Cheng, F.; Qiu, J. Internal polyadenylation of the parvovirus B19 precursor mRNA is regulated by alternative splicing. J. Biol. Chem. 2011, 286, 24793–24805.
- Guan, W.; Cheng, F.; Huang, Q.; Kleiboeker, S.; Qiu, J. Inclusion of the central exon of parvovirus B19 precursor mRNA is determined by multiple splicing enhancers in both the exon and the downstream intron. J. Virol. 2011, 85, 2463–2468.
- Bonvicini, F.; Filippone, C.; Delbarba, S.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Parvovirus B19 genome as a single, two-state replicative and transcriptional unit. Virology 2006, 347, 447–454.
- Bonvicini, F.; Filippone, C.; Manaresi, E.; Zerbini, M.; Musiani, M.; Gallinella, G. Functional analysis and quantitative determination of the expression profile of human parvovirus B19. Virology 2008, 381, 168–177.
- Bua, G.; Manaresi, E.; Bonvicini, F.; Gallinella, G. Parvovirus B19 Replication and Expression in Differentiating Erythroid Progenitor Cells. PLoS ONE 2016, 11, e0148547.
- Xu, P.; Ganaie, S.S.; Wang, X.; Wang, Z.; Kleiboeker, S.; Horton, N.C.; Heier, R.F.; Meyers, M.J.; Tavis, J.E.; Qiu, J. Endonuclease Activity Inhibition of the NS1 Protein of Parvovirus B19 as a Novel Target for Antiviral Drug Development. Antimicrob. Agents Chemother. 2019, 63, e01879-18.
- Ning, K.; Roy, A.; Cheng, F.; Xu, P.; Kleiboeker, S.; Escalante, C.R.; Wang, J.; Qiu, J. High throughput screening identifies inhibitors for parvovirus B19 infection of human erythroid progenitor cells. J. Virol. 2021, JVI0132621.
- Mietzsch, M.; Penzes, J.J.; Agbandje-McKenna, M. Twenty-Five Years of Structural Parvovirology. Viruses 2019, 11, 362.
- Ros, C.; Bieri, J.; Leisi, R. The VP1u of Human Parvovirus B19: A Multifunctional Capsid Protein with Biotechnological Applications. Viruses 2020, 12, 1463.
- Zou, W.; Ning, K.; Xu, P.; Deng, X.; Cheng, F.; Kleiboeker, S.; Qiu, J. The N-terminal 5–68 amino acids domain of the minor capsid protein VP1 of human parvovirus B19 enters human erythroid progenitors and inhibits B19 infection. J. Virol. 2021, 95, e00466-21.
- Zhi, N.; Zadori, Z.; Brown, K.E.; Tijssen, P. Construction and sequencing of an infectious clone of the human parvovirus B19. Virology 2004, 318, 142–152.
- Zhi, N.; Mills, I.P.; Lu, J.; Wong, S.; Filippone, C.; Brown, K.E. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J. Virol. 2006, 80, 5941–5950.
- Manaresi, E.; Conti, I.; Bua, G.; Bonvicini, F.; Gallinella, G. A Parvovirus B19 synthetic genome: Sequence features and functional competence. Virology 2017, 508, 54–62.
More
Information
Subjects:
Virology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
812
Revisions:
2 times
(View History)
Update Date:
17 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No