Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Saverio Alberti | + 4231 word(s) | 4231 | 2022-01-05 07:42:18 | | | |
| 2 | Camila Xu | Meta information modification | 4231 | 2022-01-13 02:57:44 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Alberti, S.; Guerra, E.; Di Pietro, R.; Trerotola, M.; Basile, M. Cancer-Homing CAR-T Cells. Encyclopedia. Available online: https://encyclopedia.pub/entry/18171 (accessed on 25 July 2026).
Alberti S, Guerra E, Di Pietro R, Trerotola M, Basile M. Cancer-Homing CAR-T Cells. Encyclopedia. Available at: https://encyclopedia.pub/entry/18171. Accessed July 25, 2026.
Alberti, Saverio, Emanuela Guerra, Roberta Di Pietro, Marco Trerotola, Mariangela Basile. "Cancer-Homing CAR-T Cells" Encyclopedia, https://encyclopedia.pub/entry/18171 (accessed July 25, 2026).
Alberti, S., Guerra, E., Di Pietro, R., Trerotola, M., & Basile, M. (2022, January 13). Cancer-Homing CAR-T Cells. In Encyclopedia. https://encyclopedia.pub/entry/18171
Alberti, Saverio, et al. "Cancer-Homing CAR-T Cells." Encyclopedia. Web. 13 January, 2022.
Copy Citation
Chimeric antigen receptor (CAR) therapy is based on patient blood-derived T cells and natural killer cells, which are engineered in vitro to recognize a target antigen in cancer cells. Most CAR-T recognize target antigens through immunoglobulin antigen-binding regions.
CAR-T cells
immune cell populations
signaling
immune checkpoint blockade
1. Introduction
Chimeric antigen receptor (CAR) therapy is based on patients’ blood-derived T cells and natural killer (NK) cells, which are engineered in vitro to express artificial receptors to recognize a specific target antigen and the cells expressing that antigen (Figure 1) [1].
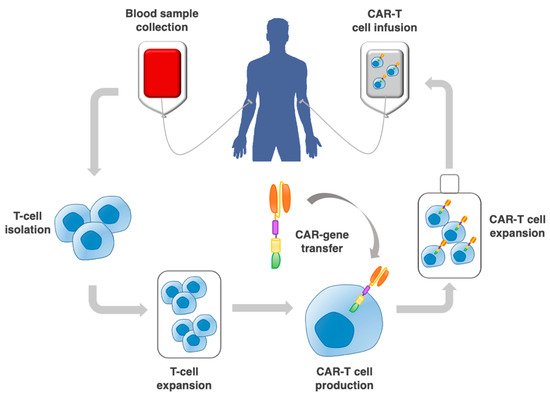
Figure 1. Procedure to implement adoptive CAR-T cell therapy. This includes the collection of a patient blood sample; T cell selection by leukapheresis from peripheral blood; CAR–gene transfer through a vector; expansion of CAR-T cells in vitro, and re-infusion into the patient.
The genetic modification of peripherally derived T cells with CAR was developed from the concept of adoptive immunotherapy using tumor-infiltrating lymphocytes (TIL) [2], whose T cell receptors (TCR) can recognize tumor-associated antigens. TCR require the presentation of processed antigens by the major histocompatibility complex (MHC). This led to the suggestion that T cells TCR with high affinity for MHC/peptide, whether of syngeneic or allogeneic origin, may be utilized for efficient anticancer immunotherapy [3].
CAR-T cells were instead developed to recognize the target antigen through immunoglobulin antigen-binding regions. Hence, CAR-T do not require target-peptide presentation by MHC molecules. This correspondingly allowed CAR constructs to be used to direct the activity of NK cells [4].
CAR-T cell therapy has been tremendously successful in the treatment of leukemias [5][6][7][8], seminal to this success being the development of anti-CD19 CAR-T. CAR-T immunotherapy has subsequently undergone intense testing for application to the treatment of solid tumors [9][10]. However, the clinical efficacy of CAR-T cells observed in hematological malignancies is rarely found against solid tumors.
CAR-T cell therapy for solid tumors faces many hurdles [9][10], starting from the very first step of intravenous administration of activated CAR-T cells, wherein in vitro engineered cells must be driven to the tumor by appropriate molecular signals. Correspondingly, early (≤ 3 days) intratumoral infiltration of CAR-T cells post-infusion was shown to be a predictor of survival, and lymphokines/cytokines that increase homing to the tumor were shown to be essential for CAR-T cell therapy outcome [11]. Additional challenges for CAR-T cells are provided by the requirement of long-term persistence within the tumor, resistance to exhaustion, and distinct interaction with other cancer infiltrating cell populations, within the largely immunosuppressive cancer environment [12]. The induction of immunosuppression was assessed using CAR targeting mesothelin or fibroblast activation protein and assessing the functional capacity of CAR-T lymphocytes infiltrating a tumor (CAR-TIL). CAR-TIL underwent rapid loss of functional activity in the tumor. This hypofunction was reversible when the T cells were isolated away from the tumor [13]. Among the factors associated with CAR-T cell hypofunction, there are the upregulation of intrinsic T-cell inhibitory enzymes (diacylglycerol kinase and SHP-1) and expression of surface inhibitory receptors (PD-1-programmed cell death protein-1, LAG3-lymphocyte-activation gene 3, TIM-3-T-cell immunoglobulin and mucin-domain containing-3, and 2B4). This confirms the immunosuppressive impact of tumor environment and shows that CAR-T cell inactivation is reversible, suggesting the feasibility of systemic approaches to overcome this tumor-induced inhibition, which include PD-1 pathway antagonism [13].
Intense efforts have gone into tackling these pitfalls. However, we argue that some CAR-engineering strategies may risk missing the big picture, i.e., that a successful CAR-T-cell therapy must efficiently intertwine with the complex and heterogeneous responses that the body has already mounted against the tumor [14]. Experimental evidence has been recently obtained that lends support to this model.
2. CAR Design
2.1. Early CAR
The concept of CAR was first proposed by Kuwana et al. in 1987 [15]. A CAR typically comprises an antibody fragment, such as an scFv or Fab fragment, incorporated in a fusion protein that contains additional components, such as a CD3ζ and a CD28 transmembrane domain, together with selective T-cell activating moieties, including the endodomains of CD28, OX40, 4-IBB, Lek, and ICOS (Figure 2) [8].
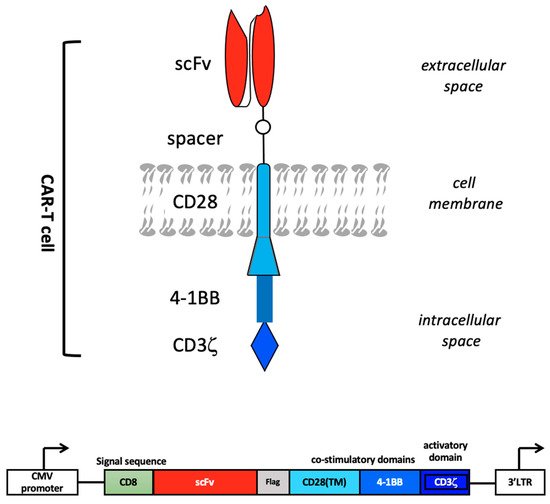
Figure 2. CAR design. The extracellular portion of the CAR molecule is typically derived from a monoclonal antibody that recognizes a cancer-associated antigen. The variable heavy (VH) and light (VL) chains, or single-chain variable fragment (scFv), are connected by a hinge to form the antigen-binding region of the CAR molecule. The antigen-binding region is linked through a transmembrane domain to an intracellular T-cell signaling domain, in particular CD3ζ, which is the primary activation domain for TCR mediated T-cell activation. A CAR construct further comprises one or more co-stimulatory domains, e.g., those derived from 4-1BB and CD28. Bottom: schematic representation of a CAR construct inserted in an expression vector, e.g., a lentiviral vector. Vector-derived CMV (cytomegalovirus)-promoter and 3′LTR (long terminal repeat) regions are depicted.
The first-generation CAR construct comprised a CD3ζ chain as a key transmitter of signals from endogenous TCR. However, cocd47-stimulation of the TCR is required for the efficient recognition of an MHC–antigen complex in a physiological TCR context. Hence, improvements in the CAR design included secondary and tertiary intracellular signaling chains, that enhanced activatory signaling. These subsequent generations of CAR, with the addition of one versus two co-stimulatory domains (in 2nd and 3rd generation CAR, respectively) have shown enhanced activity, persistence, and efficacy (Figure 3) [12].
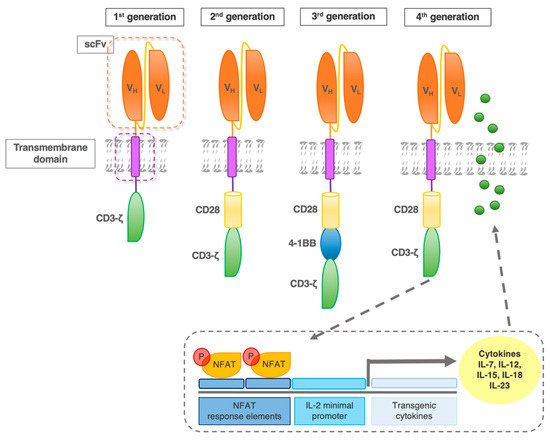
Figure 3. CAR evolution. First-generation CAR contains one intracellular signaling domain (CD3ζ) of the T cell receptor complex. Second-generation receptors contain one additional co-stimulatory domain (e.g., CD28). Based on the most effective second-generation CAR, the third generation added another co-stimulatory molecule, such as 4-1BB. Fourth-generation CAR were designed to activate NFAT (nuclear factor of activated T cells) transcription factor-driven cytokine production. After the CAR recognizes the target antigens, CD3ζ mediates downstream signaling and the activation/phosphorylation of NFAT. P-NFAT is shuttled into the nucleus and binds to the NFAT response elements/interleukin(IL)-2 minimal promoter of a transgenic expression cassette co-transfected with the CAR construct.
First-generation CAR-T entered clinical trials for leukemia, lymphoma, and various other types of cancer, including ovarian cancer and neuroblastoma [1]. Lack of activation resulted in ineffective anti-solid tumor action, but persistent exposure to the tumor environment led to some continued therapeutic effects in patients with B-cell lymphoma and neuroblastoma [1][16].
Second-generation CAR constructs [6] provided a dual signaling function for T-cell activation. The second-generation CAR aimed at integrating intracellular signaling domains from several co-stimulatory molecules, such as CD28, 4-1BB, or CD137, inducible T cell co-stimulator (ICOS) or CD278, OX40, or CD134 fused to the cytoplasmic tail of the CAR, thus amplifying CAR signaling [17]. A large degree of intergenerational variety exists between second- and third-generation CAR, with a range of co-stimulatory domains being tested (CD28, 4-1BB, OX40, CD27, ICOS, DAP10, and LAT-linker for activation of T cells) [12]. The attributes of each co-stimulatory domain differ in their ability to confer cytokine secretion, cytotoxicity, proliferation, and memory development to the modified-CAR-T cells [18].
Third-generation CAR comprised three or more signaling functions, typically incorporating CD28 transmembrane and endodomains, fused to the signaling subunits of 4-1BB, OX40, or Lek, and to the cytoplasmic domain of CD3ζ. A third-generation CAR consisting of αCD19-CD3ζ-CD28-4-1BB was shown to lead to complete remissions in patients with chronic lymphocytic leukemia [19].
2.2. Fourth-Generation CAR
Co-stimulation of TIL with cytokines such as IL-2 and IL-12 had previously been utilized to enhance anticancer responses [20]. A corresponding benefit was obtained by adding immune checkpoint blockers (ICB) [21]. The vast use of ICB in anticancer therapy in recent years has allowed to unravel distinct mechanistic aspects in adoptive immunotherapy [22] and is now helping to shed light on corresponding issue on CAR-T cell therapy. Consistently, PD-1 was shown to be upregulated on exhausted CAR-T cells and TIL, leading to loss-of-function T lymphocytes. The enhanced efficacy of CAR-T cells was correspondingly obtained by adding ICB to CAR-T cell therapy [23]. Additional insight was provided by exploiting the cancer-immune system liaison using bispecific antibodies [24]. Altogether, these approaches indicated that making use of cancer-infiltrating immune cells, while tilting the balance of immune regulatory circuits toward activation, was required to reach efficacy. Following these principles, to induce a pro-activatory milieu, CAR-T cells were engineered to release transgenic cytokines upon CAR signaling in the targeted tumor tissue. Such ‘T cells redirected for antigen-unrestricted cytokine-initiated killing’ (TRUCK) are also called ‘fourth-generation’ CAR-T cells. Through CAR-induced release, cytokines were secreted in the target tissue rather than in the circulation, thus alleviating systemic side effects. The TRUCK concept is currently being explored using a panel of cytokines, such as IL-7, IL-12, IL-15, IL-18, IL-23 (Figure 4), and combinations thereof [25]. The secretion of IL-15 or IL-18 enhances T cell proliferation. The combination of CCL19 and IL-7 recruits endogenous immune cells and establishes a memory response against tumor cells [26]. CAR-T cells engineered to secrete IL-12 have been shown to remodel the tumor microenvironment (TME) by reprogramming tumor-associated macrophages (TAM) to an M1 phenotype and decreasing the presence of myeloid-derived suppressor cells (MDSC) and Treg in syngeneic mouse models [27]. Similarly, CAR-T cells that constitutively secrete IL-18 increase intratumoral M1 macrophages, activated dendritic cells (DC), and activated NK cells numbers, while decreasing M2 macrophages and Treg (regulatory T cells) levels. A direct comparison of CAR-T cells expressing IL-12 to IL-18 indicated that IL-18 is more effective at remodeling the immunosuppressive TME in a syngeneic murine pancreatic cancer model [28].
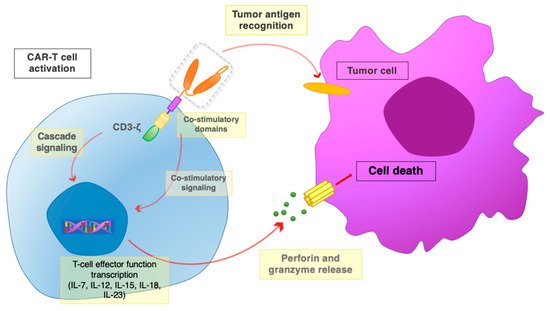
Figure 4. The CAR-T interaction with tumor cells. Once the scFv portion recognizes and binds the tumor antigen, the co-stimulatory domains and the CD3ζ chain promote activatory signaling cascades. Downstream signaling leads to the activation of T cell effector functions, with the release of perforin and granzyme, leading to the death of target tumor cells.
CAR-T cells engineered to secrete interleukin(IL)-12 stimulate innate immune cells against the tumor and become resistant to Treg and MDSC [29]. T cells were engineered with the IL-23 p40 receptor chain that associates with the endogenous IL23a p19 receptor to use released IL-23 in an autocrine fashion. CAR-T cells with engineered IL-23 receptor showed superior reactivity in various tumor models and attenuated side effects, suggesting an advantageous use of IL-23 for sustaining an improved CAR-T cell response [30].
TRUCK approaches are being tested in early phase clinical trials. The NCT03182816 trial was started in 2017 as a phase I/II single-arm to determine the safety and efficacy of infusion of autologous T cells engineered to express immune checkpoint antibodies (CTLA-4 and PD-1) and chimeric antigen receptor targeting epidermal growth factor receptor (EGFR-CAR) in adult patients with EGFR-positive advanced malignant solid tumors. Eight of nine patients showed detectable EGFR-CAR-T cells in their peripheral blood. One patient had a partial response, which lasted >13 months, six had stable disease, and two had disease progression [31]. Koneru et al. assessed in a phase I clinical trial IL-12-secreting CAR-T cells directed against MUC-16(ecto) for recurrent ovarian cancer [32]. The EGFR-IL-12-CAR-T NCT03542799 clinical trial started enrolling patients with metastatic colorectal cancer in 2018. This phase I/II study is expected to accrue 20 patients by 2021. The NCT03932565 phase I clinical trial is assessing CAR-T cells directed against Nectin4_FAP since 2019. CAR-T cells were engineered to express IL-7 and CCL19 (C-C Motif Chemokine Ligand 19) [26] or IL-12. The accrual of 30 patients is expected to be completed by 2021.
3. Main Challenges for CAR-T Cell Therapy against Solid Tumors
As mentioned above, the clinical efficacy of CAR-T cells observed in hematological malignancies is rarely found against solid tumors. Hematological malignancies are devoid of many of the physical immunosuppressive factors that hamper adoptively transferred cells from reaching solid tumors. Furthermore, target antigens that are present on hematological cancers are often homogenous and are expressed in most tumor cells. In contrast, target antigens on solid tumors are often heterogeneous both across tumors and between primary and metastatic sites. CAR-T cell therapy for solid tumors faces additional obstacles [9][10], starting from the administration route, wherein CAR-T cells injected in the peripheral blood must encounter the correct chemotactic signals to traffic to the tumor in sufficient numbers. The abnormal cancer vasculature impedes efficient passage (diapedesis) toward tumor tissues [33][34][35]. Physical barriers by hard-to-penetrate fibrotic stroma prevent adequate CAR-T cell diffusion at a distance from the blood vessels. Finally, immunosuppressive factors include inhibitory checkpoint pathway signals and immunosuppressive cytokines [36], TGF(transforming growth factor)-β among them [37][38][39]. Products of an altered metabolism, including amino acids, lipids, and DNA base precursors [40][41][42], and reactive oxygen species (ROS) [43] can be found in the tumor environment [12]. All these factors considerably affect the long-term persistence within the tumor in an active state.
3.1. CAR Target Choice for Anticancer Therapy
An appropriate choice of target antigens and the effective combination with immunostimulatory signaling have been shown to enhance CAR efficacy. CAR comprising the ICOS signaling domain liaises with the effective antitumor effect on gliomas that express the epidermal growth factor receptor variant III (EGFRvIII) [44]. The preclinical evaluation of CAR-T cell therapy targeting the tumor antigen 5T4 in ovarian cancer led to successful outcomes [45]. CAR targets included genetic products arising from gene mutations (EGFRvIII) [46], modified glycosylation patterns (MUC1) [47], cancer-testis antigen-derived peptides (MAGE), and mesothelin-specific CAR-T cells [48]. CAR was further generated that target overexpressed antigens in breast cancer, lung cancer, and pancreatic cancer, such as carcinoembryonic antigen GD2, prostate-specific membrane antigen, HER2/ERBB2, MUC16 [49] or tumor stroma (fibroblast activation protein or vascular endothelial growth factor receptor, VEGFR) [50]. Additional novel targets include the type 1 insulin-like growth factor receptor (IGF1R) and receptor tyrosine kinase-like orphan receptor 1 (ROR1) for sarcoma as well as the L1-cell adhesion molecule (L1-CAM) for ovarian cancer [51].
These combined efforts have led to a burgeoning of clinical trials in the field, the majority remaining focused, though, on leukemias and lymphomas [12]. FDA-approved CAR-T cell therapies now include CTL019 (Kymriah) [52], KTE-C19 (Yescarta) [53], JCAR017 (Breyanzi) [54], KTE-X19 (Tecartus) [55], and bb2121 (Abecma) [56].
3.2. Trafficking and Persistence of CAR-T Cells in the Tumor
The persistence, trafficking, and maintenance of function remain a challenge in many CAR-T approaches. Using intravital imaging, anti-CD19 CAR-T cells were tracked at lymphoma sites [57]. However, circulating targets trapped CAR-T cells in the lungs, reducing their access to lymphoid organs. In the bone marrow, tumor apoptosis was largely due to CAR-T cells that engaged, killed, and then detached from their targets within 25 min. Notably, not all CAR-T cell contacts elicited calcium signaling or killing while interacting with tumors, uncovering extensive functional heterogeneity. Mathematical modeling revealed that direct killing was sufficient for tumor regression. Finally, antigen-loss variants were shown to emerge in the bone marrow, but not in lymph nodes, where CAR-T cell cytotoxic activity was reduced. Hence, a previously unappreciated level of diversity exists in the outcomes of CAR-T cell interactions in distinct anatomical districts in the body, with important clinical implications [57].
3.3. CAR-T Treatment Toxicity
The anticancer activity of CAR-T can associate with life-threatening toxicity due to the increased secretion of pro-inflammatory cytokines (cytokine release syndrome, CRS) and the immune effector cell-associated neurotoxicity syndrome (ICANS) as mediated by the simultaneous activation of large numbers of T cells [8][58]. This frequently leads to multiorgan dysfunction, pulmonary failure, and death [59]. Consequent therapeutic intervention requires the intense management of treated patients [60][61].
Cytokine release is usually greater with CAR containing CD28 versus 4-1BB co-stimulatory domains. On the other hand, constructs with either domain confer similar anticancer activity in mouse models. T cell products expressing CAR with either CD28 or 4-1BB co-stimulatory domains have been highly efficacious in patients with relapsed hematological malignancies, and anti-CD19 CAR showed similar activity regardless of the source of the co-stimulatory domain. In large-cohort clinical trials, the rates of neurological toxicities have been higher with CD28–co-stimulated CAR, although this finding is probably the result of multiple converging factors rather than due to CD28 signaling alone [62]. One of these factors was the increased circulating IL-17 levels at baseline in patients with locoregional metastatic melanoma. In ICB approaches, increased IL-6 levels in patients with metastatic melanoma treated with ipilimumab (anti-CTLA-4 antibody) were shown to be associated with more intense adverse events. More global cytokine dysregulation as assessed by measuring the circulating levels of several cytokines at baseline or early on treatment has been shown to be predictive of adverse events in patients treated with anti-PD-1 therapies alone or in combination with anti-CTLA-4 therapies [21].
Intense cytokine release is associated with T-cell activation upon engagement with target cells, which leads to higher levels of circulating cytokines, including IL-6 and interferon γ. Consistently, elevated levels of serum cytokines are less common in patients who do not have a clinical response after CAR-T cell therapy [63].
NK cells produce several cytokines, including tumor necrosis factor α, interferon γ, and IL-10 [64]. NK cells produce lower levels of IL-6 than T cells [63], thus potentially reducing systemic cytokine-storm toxicity. Of note, the CAR-T cell-induced cytokine release syndrome can be mediated by macrophages and is relieved by IL-1 blockade [65].
3.4. CAR-T Cell Exhaustion
A major concern of CAR-T therapy is that CAR-T cells may become exhausted or dysfunctional [66][67], which is a phenomenon whereby the T cells become unresponsive due to overstimulation [68].
CAR-T cell exhaustion is considered the outcome of the chronic tumor stimulation imposed on T cells, which leads to disruption of their function [67]. Proof of (re-)activation of TIL upon blockade of the PD-1/PD-L1 (programmed death-ligand 1) or other checkpoint axes [69] has strongly supported this model. Correspondingly, other therapeutic measures, including adoptive T-cell therapy, epigenetic reprogramming, antibodies targeting T-cell co-stimulatory molecules, metabolic reprogramming, and conventional cancer therapies, such as chemotherapy, radiotherapy, and targeted therapy, have been implemented to enhance antitumor immunity, overcome resistance, and increase therapeutic efficacy [22].
Cell exhaustion may differentially affect T cell subpopulations, suggesting a relationship with the selection procedures utilized to manufacture CAR-T cells. Optimal cell subpopulations for adoptive cell transfer were suggested to be those that retain their memory/naïve capacities [70] to permit a greater boost in proliferation and function in vivo. Wnt signaling was shown to promote the generation of CD44(low)CD62L(high)Sca-1(high)CD122(high)Bcl-2(high) self-renewing multipotent CD8(+) memory stem cells with proliferative and antitumor capacities exceeding those of central and effector memory T cell subsets [70].
Epigenetic profiles regulate the gene expression of key transcription factors over immune cell differentiation and proliferation pathways. Through a screening of chemical probes with defined epigenetic targets, JQ1, an inhibitor of bromodomain and extra-terminal motif (BET) proteins, was found to maintain CD8+ T cells with functional properties of stem cell–like and central memory T cells [71]. Adoptively transferred in vitro-JQ1-treated CAR-T cells showed higher proliferation, persistence, and increased cytokine secretion than non-treated CAR-T cells in murine models [71].
The altered differentiation of CAR-T cells can also accompany T cell exhaustion [72]. Utilizing mesothelin-redirected CAR-T cells in pancreatic cancer, CAR dysregulation was found to be associated with a CD8+ T-to-NK-like T cell transition, as driven by SOX4 (SRY-Box Transcription Factor 4) and ID3. The downmodulation of ID3 and SOX4 expression was indicated to improve the efficacy of CAR-T cells in solid tumors by preventing or delaying T cell exhaustion [72]. These findings further reveal CAR-T cells’ plasticity as a main actor of immune response dynamics.
3.5. Counteracting Immunosuppression against CAR-T Cells
A major hurdle to be overcome by tumor-infiltrating CAR-T cells is immunosuppression. T cell exhaustion itself was shown to be an outcome of cancer-associated immunosuppression. Correspondingly, resistance to exhaustion was shown to be linked to interaction with other cancer-infiltrating cell types [12].
Cancers contain a broad ensemble of genetically normal cells within an extracellular matrix (ECM), which was collectively termed the tumor microenvironment (TME), that substantially diverges from normal stroma. This has fostered the concept that tumors grow as integrated tissues or organs, that combine diverse components, such as vasculature, nerves, an immune environment, and connective tissue.
TGF-β is a key driver of immunosuppression [37][38][39]. Prostate cancer, in particular, secretes TGF-β as a means to inhibit immunity while allowing for cancer progression. Blocking TGF-β signaling augments T cell ability to infiltrate, proliferate, and mediate antitumor responses in prostate cancer models. The potency of PSMA(prostate-specific membrane antigen)-targeted CAR-T cells was correspondingly enhanced utilizing dominant-negative TGF-βRII (dnTGF-βRII) expression in CAR-T cells [73]. This led to an increased proliferation of CAR-T cells, enhanced cytokine secretion, resistance to exhaustion, long-term in vivo persistence, and eradication of human prostate cancer in mouse models. A phase I clinical trial is being conducted to assess these CAR-T cells in relapsed and refractory metastatic prostate cancer patients. This is a non-randomized, sequential assignment, open label trial; 18 participants have been enrolled; completion of the study is expected in 2022 (ClinicalTrials.gov: NCT03089203) [73]. Knocking out the endogenous TGF-β receptor II (TGFBR2) in CAR-T cells was achieved using CRISPR/Cas9. This reduced Treg conversion prevented CAR-T cell exhaustion and achieved higher tumor eradication rates both for xenografts and for PDX (patient derived xenograft), with higher proportion of memory and effector memory CAR-T cell subsets [74]. Additional approaches were suggested to be effective, e.g., the expression of chimeric TGFBR2 and TGFBR1 where the TGF-β-binding domain is fused to the transmembrane and intracellular signaling domains of IL-12 receptor (CTBR). CAR-T/CTBR cells secreted significantly greater amounts of IFNγ than control T cells following activation in the presence of TGF-β. In the absence of IL-2, antigen-driven expansion was severely limited by exposure to TGF-β, and CAR-T cells progressively lost cytotoxic activity. Although T cells overexpressing the dominant negative TGF-β receptor failed to expand and clear tumor cells in the presence of TGF-β, CTBR expressing CAR-T cells maintained their ability to expand and kill tumor targets in the presence of TGF-β [75].
Metabolic conditions can negatively impact on CAR-T cell function: among them, an increase in the acidity of the TME because of increased glycolysis by cancer cells. This ‘Warburg effect’ [76][77] stems from a preferential utilization of glucose via glycolysis rather than via oxidative phosphorylation. Cancer-associated fibroblasts (CAF) contribute to increased intratumor glycolysis and impact on breast cancer growth [78]. High glycolysis leads also to an increase in oxidative stress and in ROS production. The secretion of high levels of ROS by MDSC contributes additional immunosuppressive capacity. Immunosuppression by oxidative stress impairs CAR-T cells proliferation and cytotoxicity [12]. This led to engineering CAR-T cells to secrete catalase (CAT), an antioxidant enzyme, into the TME. CAR-CAT-T cells were shown to regain their antitumor functions [43]. Local catalase secretion provided a bystander effect and restored cytotoxic function to NK cells.
One of the approaches used to fight immunosuppression has been the generation of CAR-T cells expressing cell-surface dominant-negative receptors (DNR) to override the inactivating signals present in the TME. DNR can be generated with a functional extra-cellular domain and a mutation in the intracellular region to abolish downstream signal transduction. DNR can effectively compete with their endogenous counterparts. The use of DNR for immunosuppressive factors such as TGF-β has endowed transduced EBV cells with resistance to immunosuppression [79]. A DNR for PD-1 on CAR-T cells rescued the effect of checkpoint blockade and restored effector functions. PD-1/PD-L1 blockade is normally achieved through systemic antibody delivery, which can result in autoimmune reactions. PD-1 ‘insensitive’ DNR T cells do not require systemic ICB and may prevent this major side effect. Switch receptors offer yet another approach to circumvent immunosuppression. These CAR contain the extracellular portion of an antibody specific for an immunosuppressive molecule, such as PD-1 or CTLA-4, which is fused to an intracellular activating signaling molecule, such as CD28. The infiltration and antitumor efficacy of PD-1-CD28 switch-CAR-T cells were enhanced versus parental CAR-T cells [80]. Switch-CAR-T cells showed a reduction in other checkpoint inhibitors, e.g., LAG3, TIM-3, and CEACAM1 (carcinoembryonic antigen-related cell adhesion molecule 1), and increased IL-2 signaling, suggesting an induction of recovery from cell exhaustion.
The reduction of inhibitory signaling pathways in T cells has shown promise in T-cell reactivation. The inhibition of Protein Kinase A with Ezrin using a ‘regulatory subunit 1 anchoring disruptor’ (RIAD-CAR) resulted in an upregulation of CXCR3 and CD49D integrin (VLA-4), which resulted in enhanced RIAD-CAR-T cells trafficking to tumors and better migration to CXCL10 in vitro [81]. RIAD-CAR cells expressed higher levels of both IFNγ and cytotoxicity and were more resistant to immunosuppression in TME.
IL-8 release within tumors was utilized to enhance intratumoral T-cell trafficking. Modified CAR inducing the expression of IL-8 receptors, CXCR1 or CXCR2, showed enhanced migration and persistence of T cells in the tumor. This induced complete tumor regression and long-lasting immunologic memory in preclinical models of glioblastoma, ovarian, and pancreatic cancer [11].
3.6. CAR-T Cells Targeting Multiple Antigens
The approaches described above supported the feasibility of ‘multi-targeted CAR-T’. The rationale was to improve selectivity for cancer cells and to reduce the off-tumor effects based on the presence or absence of two target antigens. This was also expected to lead to enhanced cytotoxicity and to reduce chances of antigen escape variants. Early work showed the synergistic effects of two individual CAR against two separate targets, i.e., folate binding protein and Her-2, together with limited antitumor efficacy in the presence of only one [84]. Dual CAR-expressing T cells were also shown to secrete higher cytokine levels than single CAR-T cells when co-cultured with dual antigen-expressing tumor targets [84].
Distinct intracellular signaling domains were also engineered onto different CAR recognizing two separate target antigens [85], both providing suboptimal activation upon binding of their target antigen. The rationale was to generate CAR-T cells that were only able to function at full capacity in the presence of both target antigens, thus limiting activation if only a single antigen was present. This concept was tested by targeting the prostate tumor antigens PSMA and PSCA (prostate stem cell antigen). Co-transduced T cells were shown to kill tumors that expressed both antigens but not tumors expressing either antigen alone [85].
References
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056.
- Rosenberg Steven, A.; Spiess, P.; Lafreniere, R. A New Approach to the Adoptive Immunotherapy of Cancer with Tumor-Infiltrating Lymphocytes. Science 1986, 233, 1318–1321.
- Alberti, S. A high affinity T cell receptor? Immunol. Cell Biol. 1996, 74, 292–297.
- Hermanson, D.L.; Kaufman, D.S. Utilizing chimeric antigen receptors to direct natural killer cell activity. Front. Immunol. 2015, 6, 195.
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells. Blood 2012, 119, 2709–2720.
- Ritchie, D.S.; Neeson, P.J.; Khot, A.; Peinert, S.; Tai, T.; Tainton, K.; Chen, K.; Shin, M.; Wall, D.M.; Honemann, D.; et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Mol. Ther. 2013, 21, 2122–2129.
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 2013, 5, 177ra38.
- Sadelain, M.; Brentjens, R.; Riviere, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398.
- Lim, W.A.; June, C.H. The Principles of Engineering Immune Cells to Treat Cancer. Cell 2017, 168, 724–740.
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365.
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016.
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363.
- Moon, E.K.; Wang, L.C.; Dolfi, D.V.; Wilson, C.B.; Ranganathan, R.; Sun, J.; Kapoor, V.; Scholler, J.; Puré, E.; Milone, M.C.; et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin. Cancer Res. 2014, 20, 4262–4273.
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488.
- Kuwana, Y.; Asakura, Y.; Utsunomiya, N.; Nakanishi, M.; Arata, Y.; Itoh, S.; Nagase, F.; Kurosawa, Y. Expression of chimeric receptor composed of immunoglobulin-derived V resions and T-cell receptor-derived C regions. Biochem. Biophys. Res. Commun. 1987, 149, 960–968.
- Duong, C.P.; Yong, C.S.; Kershaw, M.H.; Slaney, C.Y.; Darcy, P.K. Cancer immunotherapy utilizing gene-modified T cells: From the bench to the clinic. Mol. Immunol. 2015, 67, 46–57.
- Till, B.G.; Jensen, M.C.; Wang, J.; Qian, X.; Gopal, A.K.; Maloney, D.G.; Lindgren, C.G.; Lin, Y.; Pagel, J.M.; Budde, L.E.; et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: Pilot clinical trial results. Blood 2012, 119, 3940–3950.
- Kawalekar, O.U.; O’Connor, R.S.; Fraietta, J.A.; Guo, L.; McGettigan, S.E.; Posey, A.D.; Patel, P.R.; Guedan, S.; Scholler, J.; Keith, B.; et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity 2016, 44, 380–390.
- Tang, X.Y.; Sun, Y.; Zhang, A.; Hu, G.L.; Cao, W.; Wang, D.H.; Zhang, B.; Chen, H. Third-generation CD28/4-1BB chimeric antigen receptor T cells for chemotherapy relapsed or refractory acute lymphoblastic leukaemia: A non-randomised, open-label phase I trial protocol. BMJ Open 2016, 6, e013904.
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680.
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309–5337.
- Pitter, M.R.; Zou, W. Uncovering the Immunoregulatory Function and Therapeutic Potential of the PD-1/PD-L1 Axis in Cancer. Cancer Res. 2021, 81, 5141–5143.
- John, L.B.; Devaud, C.; Duong, C.P.; Yong, C.S.; Beavis, P.A.; Haynes, N.M.; Chow, M.T.; Smyth, M.J.; Kershaw, M.H.; Darcy, P.K. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 2013, 19, 5636–5646.
- Blanco, B.; Domínguez-Alonso, C.; Alvarez-Vallina, L. Bispecific Immunomodulatory Antibodies for Cancer Immunotherapy. Clin. Cancer Res. 2021, 27, 5457.
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84.
- Adachi, K.; Kano, Y.; Nagai, T.; Okuyama, N.; Sakoda, Y.; Tamada, K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018, 36, 346–351.
- Liu, Y.; Di, S.; Shi, B.; Zhang, H.; Wang, Y.; Wu, X.; Luo, H.; Wang, H.; Li, Z.; Jiang, H. Armored Inducible Expression of IL-12 Enhances Antitumor Activity of Glypican-3–Targeted Chimeric Antigen Receptor–Engineered T Cells in Hepatocellular Carcinoma. J. Immunol. 2019, 203, 198.
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219.
- Pegram, H.J.; Lee, J.C.; Hayman, E.G.; Imperato, G.H.; Tedder, T.F.; Sadelain, M.; Brentjens, R.J. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood 2012, 119, 4133–4141.
- Ma, X.; Shou, P.; Smith, C.; Chen, Y.; Du, H.; Sun, C.; Porterfield Kren, N.; Michaud, D.; Ahn, S.; Vincent, B.; et al. Interleukin-23 engineering improves CAR T cell function in solid tumors. Nat. Biotechnol. 2020, 38, 448–459.
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J.; et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734.
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102.
- Jain, R.K. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science 2005, 307, 58–62.
- Motz, G.T.; Santoro, S.P.; Wang, L.-P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615.
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63.
- Chen, Y.; McAndrews, K.M.; Kalluri, R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat. Rev. Clin. Oncol. 2021, 18, 792–804.
- David, C.J.; Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 2018, 19, 419–435.
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940.
- Tauriello, D.V.F.; Sancho, E.; Batlle, E. Overcoming TGFβ-mediated immune evasion in cancer. Nat. Rev. Cancer 2022, 22, 25–44.
- Parker, S.J.; Amendola, C.R.; Hollinshead, K.E.R.; Yu, Q.; Yamamoto, K.; Encarnación-Rosado, J.; Rose, R.E.; LaRue, M.M.; Sohn, A.S.W.; Biancur, D.E.; et al. Selective Alanine Transporter Utilization Creates a Targetable Metabolic Niche in Pancreatic Cancer. Cancer Discov. 2020, 10, 1018–1037.
- Olivares, O.; Mayers, J.R.; Gouirand, V.; Torrence, M.E.; Gicquel, T.; Borge, L.; Lac, S.; Roques, J.; Lavaut, M.-N.; Berthezène, P.; et al. Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat. Commun. 2017, 8, 16031.
- Auciello, F.R.; Bulusu, V.; Oon, C.; Tait-Mulder, J.; Berry, M.; Bhattacharyya, S.; Tumanov, S.; Allen-Petersen, B.L.; Link, J.; Kendsersky, N.D.; et al. A Stromal Lysolipid-Autotaxin Signaling Axis Promotes Pancreatic Tumor Progression. Cancer Discov. 2019, 9, 617–627.
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed Catalase Protects Chimeric Antigen Receptor-Redirected T Cells as well as Bystander Cells from Oxidative Stress-Induced Loss of Antitumor Activity. J. Immunol. 2016, 196, 759–766.
- Shen, C.J.; Yang, Y.X.; Han, E.Q.; Cao, N.; Wang, Y.F.; Wang, Y.; Zhao, Y.Y.; Zhao, L.M.; Cui, J.; Gupta, P.; et al. Chimeric antigen receptor containing ICOS signaling domain mediates specific and efficient antitumor effect of T cells against EGFRvIII expressing glioma. J. Hematol. Oncol. 2013, 6, 33.
- Owens, G.L.; Sheard, V.E.; Kalaitsidou, M.; Blount, D.; Lad, Y.; Cheadle, E.J.; Edmondson, R.J.; Kooner, G.; Gilham, D.E.; Harrop, R. Preclinical Assessment of CAR T-Cell Therapy Targeting the Tumor Antigen 5T4 in Ovarian Cancer. J. Immunother. 2018, 41, 130–140.
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9.
- Posey, A.D., Jr.; Schwab, R.D.; Boesteanu, A.C.; Steentoft, C.; Mandel, U.; Engels, B.; Stone, J.D.; Madsen, T.D.; Schreiber, K.; Haines, K.M.; et al. Engineered CAR T Cells Targeting the Cancer-Associated Tn-Glycoform of the Membrane Mucin MUC1 Control Adenocarcinoma. Immunity 2016, 44, 1444–1454.
- Zhang, Q.; Liu, G.; Liu, J.; Yang, M.; Fu, J.; Liu, G.; Li, D.; Gu, Z.; Zhang, L.; Pan, Y.; et al. The antitumor capacity of mesothelin-CAR-T cells in targeting solid tumors in mice. Mol. Ther.-Oncolytics 2021, 20, 556–568.
- Ahmed, N.; Brawley, V.S.; Hegde, M.; Robertson, C.; Ghazi, A.; Gerken, C.; Liu, E.; Dakhova, O.; Ashoori, A.; Corder, A.; et al. Human Epidermal Growth Factor Receptor 2 (HER2)-Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J. Clin. Oncol. 2015, 33, 1688–1696.
- Akce, M.; Zaidi, M.Y.; Waller, E.K.; El-Rayes, B.F.; Lesinski, G.B. The Potential of CAR T Cell Therapy in Pancreatic Cancer. Front. Immunol. 2018, 9, 2166.
- Hong, H.; Brown, C.E.; Ostberg, J.R.; Priceman, S.J.; Chang, W.C.; Weng, L.; Lin, P.; Wakabayashi, M.T.; Jensen, M.C.; Forman, S.J. L1 Cell Adhesion Molecule-Specific Chimeric Antigen Receptor-Redirected Human T Cells Exhibit Specific and Efficient Antitumor Activity against Human Ovarian Cancer in Mice. PLoS ONE 2016, 11, e0146885.
- Bach, P.B.; Giralt, S.A.; Saltz, L.B. FDA Approval of Tisagenlecleucel: Promise and Complexities of a $475000 Cancer Drug. JAMA 2017, 318, 1861–1862.
- Fala, L. Yescarta (Axicabtagene Ciloleucel) Second CAR T-Cell Therapy Approved for Patients with Certain Types of Large B-Cell Lymphoma. Am. Health Drug Benefits 2018, 11, 109–111.
- Abramson, J.S.; Palomba, M.L.; Gordon, L.I.; Lunning, M.A.; Wang, M.; Arnason, J.; Mehta, A.; Purev, E.; Maloney, D.G.; Andreadis, C.; et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): A multicentre seamless design study. Lancet 2020, 396, 839–852.
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.C.; Boissel, N.; Cassaday, R.D.; Leguay, T.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: Phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet 2021, 398, 491–502.
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716.
- Cazaux, M.; Grandjean, C.L.; Lemaitre, F.; Garcia, Z.; Beck, R.J.; Milo, I.; Postat, J.; Beltman, J.B.; Cheadle, E.J.; Bousso, P. Single-cell imaging of CAR T cell activity in vivo reveals extensive functional and anatomical heterogeneity. J. Exp. Med. 2019, 216, 1038–1049.
- Van De Vyver, A.J.; Marrer-Berger, E.; Wang, K.; Lehr, T.; Walz, A.-C. Cytokine Release Syndrome By T-cell–Redirecting Therapies: Can We Predict and Modulate Patient Risk? Clin. Cancer Res. 2021, 27, 6083.
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418.
- Sheth, V.S.; Gauthier, J. Taming the beast: CRS and ICANS after CAR T-cell therapy for ALL. Bone Marrow Transplant. 2021, 56, 552–566.
- Santomasso, B.D.; Nastoupil, L.J.; Adkins, S.; Lacchetti, C.; Schneider, B.J.; Anadkat, M.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; et al. Management of Immune-Related Adverse Events in Patients Treated With Chimeric Antigen Receptor T-Cell Therapy: ASCO Guideline. J. Clin. Oncol. 2021, 39, 3978–3992.
- Cappell, K.M.; Kochenderfer, J.N. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat. Rev. Clin. Oncol. 2021, 18, 715–727.
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273.
- Jiang, H.; Zhang, W.; Shang, P.; Zhang, H.; Fu, W.; Ye, F.; Zeng, T.; Huang, H.; Zhang, X.; Sun, W.; et al. Transfection of chimeric anti-CD138 gene enhances natural killer cell activation and killing of multiple myeloma cells. Mol. Oncol. 2014, 8, 297–310.
- Giavridis, T.; van der Stegen, S.J.C.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat. Med. 2018, 24, 731–738.
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674.
- Lopez de Rodas, M.; Schalper, K.A. Tumour antigen-induced T cell exhaustion—The archenemy of immune-hot malignancies. Nat. Rev. Clin. Oncol. 2021, 18, 749–750.
- Miggelbrink, A.M.; Jackson, J.D.; Lorrey, S.J.; Srinivasan, E.S.; Waibl-Polania, J.; Wilkinson, D.S.; Fecci, P.E. CD4 T-Cell Exhaustion: Does It Exist and What Are Its Roles in Cancer? Clin. Cancer Res. 2021, 27, 5742.
- Wang, H.; Kaur, G.; Sankin, A.I.; Chen, F.; Guan, F.; Zang, X. Immune checkpoint blockade and CAR-T cell therapy in hematologic malignancies. J. Hematol. Oncol. 2019, 12, 59.
- Gattinoni, L.; Zhong, X.S.; Palmer, D.C.; Ji, Y.; Hinrichs, C.S.; Yu, Z.; Wrzesinski, C.; Boni, A.; Cassard, L.; Garvin, L.M.; et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat. Med. 2009, 15, 808–813.
- Kagoya, Y.; Nakatsugawa, M.; Yamashita, Y.; Ochi, T.; Guo, T.; Anczurowski, M.; Saso, K.; Butler, M.O.; Arrowsmith, C.H.; Hirano, N. BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J. Clin. Investig. 2016, 126, 3479–3494.
- Good, C.R.; Aznar, M.A.; Kuramitsu, S.; Samareh, P.; Agarwal, S.; Donahue, G.; Ishiyama, K.; Wellhausen, N.; Rennels, A.K.; Ma, Y.; et al. An NK-like CAR T cell transition in CAR T cell dysfunction. Cell 2021, 184, 6081–6100.e26.
- Kloss, C.C.; Lee, J.; Zhang, A.; Chen, F.; Melenhorst, J.J.; Lacey, S.F.; Maus, M.V.; Fraietta, J.A.; Zhao, Y.; June, C.H. Dominant-Negative TGF-β Receptor Enhances PSMA-Targeted Human CAR T Cell Proliferation And Augments Prostate Cancer Eradication. Mol. Ther. 2018, 26, 1855–1866.
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977.
- Boyerinas, B.; Miller, S.M.; Murray, R.C.; Evans, J.W.; Parsons, G.B.; Seidl, K.J.; Friedman, K.M.; Morgan, R.A. A Novel TGF-β2/Interleukin Receptor Signal Conversion Platform That Protects CAR/TCR T Cells from TGF-β2-Mediated Immune Suppression and Induces T Cell Supportive Signaling Networks. Blood 2017, 130, 1911.
- Kim, J.W.; Dang, C.V. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006, 66, 8927–8930.
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The Bioenergetic Signature of Cancer: A Marker of Tumor Progression. Cancer Res. 2002, 62, 6674–6681.
- Migneco, G.; Whitaker-Menezes, D.; Chiavarina, B.; Castello-Cros, R.; Pavlides, S.; Pestell, R.G.; Fatatis, A.; Flomenberg, N.; Tsirigos, A.; Howell, A.; et al. Glycolytic cancer associated fibroblasts promote breast cancer tumor growth, without a measurable increase in angiogenesis: Evidence for stromal-epithelial metabolic coupling. Cell Cycle 2010, 9, 2412–2422.
- Bollard, C.M.; Rössig, C.; Calonge, M.J.; Huls, M.H.; Wagner, H.J.; Massague, J.; Brenner, M.K.; Heslop, H.E.; Rooney, C.M. Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 2002, 99, 3179–3187.
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590.
- Newick, K.; O’Brien, S.; Sun, J.; Kapoor, V.; Maceyko, S.; Lo, A.; Puré, E.; Moon, E.; Albelda, S.M. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol. Res. 2016, 4, 541–551.
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming Anti-EGFRvIII CAR-T With TGFβ Trap Improves Antitumor Efficacy in Glioma Mouse Models. Front. Oncol. 2020, 10, 1117.
- Durgin, J.S.; Henderson, F., Jr.; Nasrallah, M.P.; Mohan, S.; Wang, S.; Lacey, S.F.; Melenhorst, J.J.; Desai, A.S.; Lee, J.Y.K.; Maus, M.V.; et al. Case Report: Prolonged Survival Following EGFRvIII CAR T Cell Treatment for Recurrent Glioblastoma. Front. Oncol. 2021, 11, 669071.
- Duong, C.P.; Westwood, J.A.; Berry, L.J.; Darcy, P.K.; Kershaw, M.H. Enhancing the specificity of T-cell cultures for adoptive immunotherapy of cancer. Immunotherapy 2011, 3, 33–48.
- Kloss, C.C.; Condomines, M.; Cartellieri, M.; Bachmann, M.; Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 2013, 31, 71–75.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.4K
Revisions:
2 times
(View History)
Update Date:
13 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No