Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Miranda Fernández-Serrano | + 1730 word(s) | 1730 | 2022-01-03 08:13:33 | | | |
| 2 | Conner Chen | Meta information modification | 1730 | 2022-01-13 06:53:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Fernández-Serrano, M. B-cell non-Hodgkin lymphoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/18160 (accessed on 23 July 2026).
Fernández-Serrano M. B-cell non-Hodgkin lymphoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/18160. Accessed July 23, 2026.
Fernández-Serrano, Miranda. "B-cell non-Hodgkin lymphoma" Encyclopedia, https://encyclopedia.pub/entry/18160 (accessed July 23, 2026).
Fernández-Serrano, M. (2022, January 12). B-cell non-Hodgkin lymphoma. In Encyclopedia. https://encyclopedia.pub/entry/18160
Fernández-Serrano, Miranda. "B-cell non-Hodgkin lymphoma." Encyclopedia. Web. 12 January, 2022.
Copy Citation
The term B-cell non-Hodgkin lymphoma (B-NHL) encompasses different neoplasms characterized by an abnormal proliferation of lymphoid B cells.
non-Hodgkin lymphoma
B-cell lymphoma
epigenetics
DNA methylation
DNMT
HAT
HDAC
EZH2
PRMT
HMT
HDMT
bromodomain inhibitors
1. Characteristics of the Main B-NHL Subtypes
B-NHL are sub-divided into distinct categories based on the differentiation stage of the aberrant B cell and the presence of specific genetic alterations.
Diffuse large B-cell lymphoma (DLBCL) is the most common subtype of B-NHL, accounting for 25–35% of all cases. Although different genetic subsets with distinct genotypic, epigenetic, and clinical characteristics have been recently identified by high-throughput sequencing [1][2], the three molecular subtypes defined in the early 2000s by gene expression profiling, namely germinal center B-cell (GCB)-like, activated B-cell (ABC)-like, and unclassifiable [3], are still widely used in clinics. Patients with ABC-DLBCL or genetic alterations in MYC and BCL2 and/or BCL6, called double hit (DHL) or triple hit lymphomas (THL), generally have a poor survival prognosis. The standard of care for DLBCL is an immunochemotherapeutic regimen combining the chemotherapeutic drugs cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) with the anti-CD20 monoclonal antibody rituximab (R-CHOP) [4][5].
Follicular lymphoma (FL) is a neoplasm originating from germinal center (GC) cells with a follicular pattern. It is the second most common B-NHL, accounting for 20% of all B-NHL cases. FL is characterized by the t(14;18)(q32;q21) translocation involving the BCL2 gene, present in 90% of grade 1–2 patients [6][7]. However, the clinical course is mostly indolent; about 20% of patients, despite treatment, relapse or progress to transformed-FL (t-FL), a more aggressive subtype. Treatment usually involves localized radiotherapy for early stages and rituximab combined with chemotherapy regimens like CHOP for advanced stages [7][8].
Burkitt lymphoma (BL) is another GC-derived lymphoma characterized by the deregulation of MYC due to translocations such as t(8;14)(q23;q32). Three subtypes have been described, namely endemic, sporadic, and immunodeficiency-associated form, which is mostly found in patients infected with the human immunodeficiency virus (HIV). Although BL is an aggressive neoplasm, most patients respond to intensive chemotherapeutic regimens [9][10].
Marginal zone lymphomas (MZL), accounting for 5–15% of B-NHL, originate from memory B cells. The three described clinical entities are splenic (SMZL), nodal (NMZL), and extra-nodal MZL (EMZL), arising from the marginal zone of the spleen, the lymph nodes, and the mucosa-associated lymphoid tissue (MALT), respectively. EMZL, the most common subtype, is associated with chronic inflammation, such as that derived from Helicobacter pylori infections [11][12][13]. Clinical evolution is mostly slow. Treatment usually involves antibiotic treatment for H. pylori-positive gastric EMZL, splenectomy for SMZL, radiotherapy for localized disease, and chemotherapy regimens combined with rituximab for advanced stages [14].
Mantle cell lymphoma (MCL) originates from mature B cells in the mantle zone of lymph nodes and accounts for 3–10% of B-NHL. Its molecular hallmark is the t(11;14)(q13;q32) translocation, which leads to the overexpression of cyclin D1 (CCND1). MCL has a poor prognosis due to diagnosis often at a disseminated stage and an aggressive clinical evolution. Treatment usually involves immunochemotherapy regimens such as R-CHOP, followed by rituximab maintenance, and autologous stem cell transplantation (ASCT) in fit cases [15][16] (Figure 1).
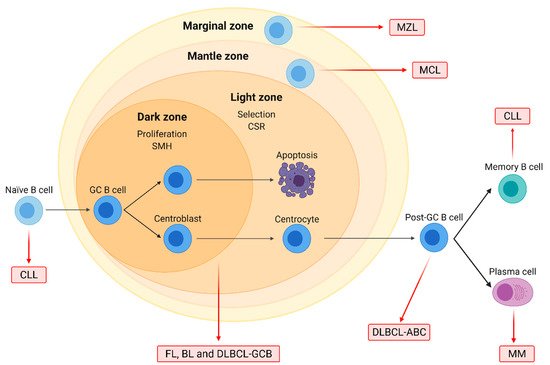
Figure 1. Origin of the major B-cell non-Hodgkin lymphoma (B-NHL) subtypes. Naïve B-cells form germinal centers (GC) after interacting with antigens. In the dark zone, centroblasts proliferate and undergo somatic hypermutation (SMH), while in the light zone, centrocytes are selected based on BCR affinity and undergo class-switch recombination (CSR). GC cells are the normal counterparts of follicular lymphoma (FL), Burkitt lymphoma (BL), and diffuse large B-cell lymphoma (DLBCL) of the GC subtype (GCB). DLBCL of the activated B-cell (ABC) subtype originates from post-GC cells, and multiple myeloma (MM) arises from differentiated plasma cells. Chronic lymphocytic leukemia (CLL) may originate from either naïve or differentiated memory B cells. Mantle cell (MCL) and marginal zone lymphoma (MZL) arise from B cells located on the mantle and the marginal zone of lymphoid follicles, respectively.
In conclusion, B-NHL subtypes range in their severity from well-controlled indolent diseases to extremely aggressive forms that have an unmet need for the development of novel therapeutic options. Related B-cell-derived neoplasms further include multiple myeloma (MM) and chronic lymphocytic leukemia (CLL).
2. Epigenetic Modification of Histone Proteins in B-NHL
Epigenetics was defined in 1942 by Conrad H. Waddington as “the branch of biology which studies the causal interactions between genes and their products, which bring the phenotype into being” [17]. The more scientific knowledge evolved, the more epigenetics became understood as a group of molecular mechanisms constituting a level of memory of previous signals by marking genomic loci and determining the accessibility of embedded genes [18]. Thus, epigenetics is the bridge between the genotype and the phenotype, anchored in the structure and packaging of the genome into chromatin.
The chromatin inside the nucleic compartment is highly compacted and formed by RNA, DNA, and proteins. Importantly, chromatin has a three-dimensional structure that is dynamic and varies, not only between cells of the same or different cell types, but also during the lifespan of a cell itself [19]. Within the interphase nucleus, the chromatin is present as chromosomes, which occupy separate and distinct spaces denominated chromosome territories [20]. Differences in the state of compaction are visible when staining chromatin; less dense regions indicate more loosely packed euchromatin enriched in active transcription, while the strongly stained regions indicate denser heterochromatin harboring repressed genes and tandem repeats such as microsatellites, minisatellites, and transposons [21]. Heterochromatin is frequently enriched at the periphery of the nucleus and on the surface of the nucleolus. Molecular mechanisms exist to transform euchromatin into heterochromatin and vice versa, which allows genes to be expressed differently based on cell type and differentiation state.
The nucleosome is the structural unit of chromatin. The positioning of nucleosomes and their density is the first level of chromatin compaction [22]. In more recent years, attention has been drawn to chromatin motion as a separate phenomenon from compaction status [23]. Nucleosomes consist of 146 pairs of nucleotides wrapped in two loops around an octamer of 8 core histone proteins [24]. More specifically, each nucleosome contains two H2A histones, two H2B histones, two H3 histones, and two H4 histones. Histone H1 is not part of the nucleosome but stabilizes chromatin between nucleosomes to achieve a higher level of structure [25].
Several epigenetic mechanisms operate on the level of the nucleosome. Histone variants can replace replication-coupled histones and provide the nucleosome with different biochemical and biophysical properties [26]. The N-terminal and also some C-terminal tails of histones protrude out of the compact structure of the nucleosome [24] and serve as a platform for many post-translational modifications (PTMs) [27]. PTMs can also occur on the core histone fold, where they directly affect the histone-DNA interaction [28]. Many types of histones PTMs exist that include methylation, acetylation, ubiquitination, SUMOylation, citrullination, glycosylation, ADP-ribosylation, and phosphorylation [27]. The combinatorial nature of PTMs at histone residues led to the controversial hypothesis from Strahl and Allis of a “histone code” on top of the genetic code [29]. Most histone PTMs are catalyzed by enzymes. In the jargon of the chromatin community, we talk about these enzymes as “writers” of PTMs that are recognized by “readers” and removed by “erasers” [27]. The dynamic nature of these mechanisms and their interactions with the transcriptional machinery provide robustness to gene expression programs and a memory of extrinsic or intrinsic stimuli, thereby contributing to the identity and fate of a cell. This precise regulation is perturbed in cancer [30]. The relation with the transcriptional regulation is best understood for the mutual exclusive acetylation and methylation of lysine residues [31].
The writers and erasers of acetylation are histone acetyltransferases (HATs) and histone deacetylase (HDACs), respectively. HATs can be divided into several families: the GNAT family with GCN5; the MYST family including MOZ and Tip60; and the p300/CBP family, among others [32]. Similarly, HDACs are separated into three classes that have zinc-dependent enzymatic activity [33] and the Sirtuins that are NAD+-dependent [34]. The transfer of a methyl group from S-adenosyl methionine (SAM) to a lysine or arginine residue is facilitated by histone methyl transferases (HMTs) such as G9a, EZH2, or protein arginine methyltransferases (PRMTs) [35]. The corresponding erasers are histone demethylases (HDMs), of which LSD1 is an important example [35].
Independent from histones, cytosine methylation is a repressive mark occurring on the DNA molecule. DNA methyltransferases (DNMTs) add methyl groups to cytosine bases [36]. Importantly, DNA methylation can only be indirectly removed by TET enzymes and subsequent DNA repair pathways or by dilution through cell divisions [37].
Genes encoding chromatin-modifying enzymes are frequently mutated in B-cell lymphomas. In DLBCL patients, recurrent mutations affect the writers CBP (also named CREBBP), EP300, EZH2, DNMT3A, or KMT2D/MLL4, among others [38]. Chromatin-modifying enzymes can also be strongly overexpressed in B-NHL subtypes, for instance, the arginine methyltransferase PRMT5 in DLBCL and MCL [39]. Furthermore, oncogenic drivers of B-NHL, such as MYC and BCL-6, act as recruitment platforms for chromatin-modifying enzymes resulting in an altered epigenetic landscape. MYC interacts with the acetylation writers p300, GCN5, and Tip60; the erasers HDAC1 and HDAC3; the histone demethylases LSD1 and KDM4B; and the DNA methyltransferase DNMT3A [40]. BCL-6 can recruit CBP, class I, and II HDACs as well as components of the nucleosome remodeling NuRD complex or polycomb proteins [41][42][43][44] (Figure 2).
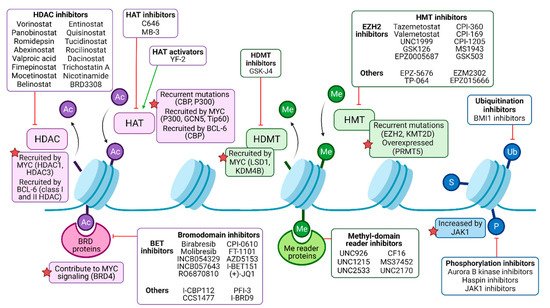
Figure 2. Pharmacological modulation of deregulated histone modifiers in B-NHL. Histone acetylation (Ac) is catalyzed by histone acetyltransferases (HATs), frequently recruited by oncogenic drivers MYC and/or BCL-6 in malignant B cells, and may be targeted by either activators or inhibitors. Histone deacetylases (HDACs) mediate deacetylation and are the target of numerous inhibitory drugs, as some of them present recurrent mutations and B-NHL and may be recruited by MYC and/or BCL-6 as well. Bromodomain (BRD)-containing proteins can bind to acetylated residues, enhancing oncogenic signaling (such as MYC program, in the case of BRD4), and can be targeted by pan or isoform-specific inhibitors. Methylation (Me) is regulated by histone methyltransferases (HMTs) and histone demethylases (HDMTs). HMTs are common targets of epigenetic drugs, especially the EZH2 subunit of the polycomb repressor complex 2 (PRC2), since it is recurrently mutated in B-NHL. HDMT inhibitors can partially counteract MYC signaling, among other effects. Me domain reader proteins, such as those containing chromodomains, may be targeted by inhibitors as well. Other histone modifications include ubiquitination (Ub), sumoylation (S), and phosphorylation (P), but their role in B-NHL pathogenesis and their targeting require further studies.
In conclusion, mechanisms of epigenetic regulation are disrupted in B-NHL through mutations, overexpression, and false recruitment of chromatin-modifying enzymes. By targeting chromatin-modifying enzymes with epidrugs, the field aims at reverting epigenetic changes on chromatin for therapy [45]. The oldest epidrugs are azanucleosides that inhibit DNMT enzymes [46].
References
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679-690.
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396-1407.
- Alizadeh, A.A.; Elsen, M.B.; Davis, R.E.; Ma, C.L.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503-511.
- Gascoyne, R.D.; Campo, E.; Jaffe, E.S.; Chan, W.C. Diffuse Large B-Cell Lymphoma, NOS. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 291–297.
- Liu, Y.; Barta, S.K.; Diffuse large B-cell lymphoma: 2019 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2019, 94, 604-616.
- Jaffe, E.S.; Harris, N.L.; Swerdlow, S.H.; Ott, G.; Nathwani, B.N.; de Jong, D.; Yoshino, T.; Spagnolo, D.; Gascoyne, R.D.. Follicular lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 266-277.
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J.; Follicular lymphoma. Nat. Rev. Dis. Prim. 2019, 5, 83.
- Dreyling, M.; Ghielmini, M.; Rule, S.; Salles, G.A.; Vitolo, U.; Ladetto, M.; Newly diagnosed and relapsed follicular lymphoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, v83-v90.
- Leoncini, L.; Campo, E.; Stein, H.; Harris, N.L.; Jaffe, E.S.; Kluin, P.M.. Burkitt Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 330-334.
- Casulo, C.; Friedberg, J.W.; Burkitt lymphoma- a rare but challenging lymphoma. Best. Pract. Res. Clin. Haematol. 2018, 31, 279-284.
- Piris, M.A.; Isaacson, P.G.; Swerdlow, S.H.; Thieblemont, C.; Pittaluga, S.; Rossi, D.; Harris, N.L.. Splenic Marginal Zone Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 223-225.
- Campo, E.; Pileri, S.A.; Jaffe, E.S.; Nathwani, B.N.; Stein, H.; Müller-Hermelink, H.K.. Nodal Marginal Zone Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 263–265.
- Cook, J.R.; Isaacson, P.G.; Chott, A.; Nakamura, S.; Müller-Hermelink, H.K.; Harris, N.L.; Swerdlow, S.H.. Extranodal Marginal Zone Lymphoma of Mucosa-Associated Lymphoid Tissue (MALT Lymphoma). In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 259–262.
- Sindel, A.; Al-Juhaishi, T.; Yazbeck, V.; Marginal Zone Lymphoma: State-of-the-Art Treatment. Curr. Treat. Options Oncol. 2019, 20, 90.
- Swerdlow, S.H.; Campo, E.; Seto, M.; Müller-Hermelink, H.K.. Mantle Cell Lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer (IARC): Lyon, France, 2017; pp. 285-290.
- Roué, G.; Sola, B.; Management of drug resistance in mantle cell lymphoma. Cancers 2020, 12, 1565.
- Waddington, C.H.; The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10-13.
- Spitz, F.; Furlong, E.E.M.; Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613-626.
- Hauer, M.H.; Gasser, S.M.; Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204-2221.
- Misteli, T.; The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28-45.
- Hildebrand, E.M.; Dekker, J.; Mechanisms and Functions of Chromosome Compartmentalization. Trends Biochem. Sci. 2020, 45, 385-396.
- Jiang, C.; Pugh, B.F.; Nucleosome positioning and gene regulation: Advances through genomics. Nat. Rev. Genet. 2009, 10, 161-172.
- Shaban, H.A.; Barth, R.; Recoules, L.; Bystricky, K.; Nanoscale mapping of nuclear dynamics in single living cells. Genome Biol. 2020, 21, 1-21.
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J.; Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251-260.
- Garcia-Saez, I.; Menoni, H.; Boopathi, R.; Shukla, M.S.; Soueidan, L.; Noirclerc-Savoye, M.; Le Roy, A.; Skoufias, D.A.; Bednar, J.; Hamiche, A.; et al. tructure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol. Cell 2018, 72, 902-915.e7.
- Buschbeck, M.; Hake, S.B.; Variants of core histones and their roles in cell fate decisions, development and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 299-314.
- Tessarz, P.; Kouzarides, T.; Histone core modifications regulating nucleosome structure and dynamics. Nat. Rev. Mol. Cell Biol. 2014, 15, 703-708.
- Tropberger, P.; Schneider, R.; Scratching the (lateral) surface of chromatin regulation by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 657-661.
- Strahl, B.D.; Allis, C.D.; The language of covalent histone modifications.. Nature 2000, 403, 41-45.
- Audia, J.E.; Campbell, R.M.; Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, 1-32.
- Morgan, M.A.J.; Shilatifard, A.; Reevaluating the roles of histone-modifying enzymes and their associated chromatin modifications in transcriptional regulation. Nat. Genet. 2020, 52, 1271-1281.
- Roth, S.Y.; Denu, J.M.; Allis, C.D.; Histone Acetyltransferases. Annu. Rev. Biochem. 2001, 70, 81-120.
- Barneda-Zahonero, B.; Parra, M.; Histone deacetylases and cancer. Mol. Oncol. 2012, 6, 579-589.
- Bosch-Presegué, L.; Vaquero, A.; Sirtuin-dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015, 282, 1745-1767.
- Morera, L.; Lübbert, M.; Jung, M.; Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 16.
- Blecua, P.; Martinez-Verbo, L.; Esteller, M.; The DNA methylation landscape of hematological malignancies: An update. Mol. Oncol. 2020, 14, 1616-1639.
- Rasmussen, K.D.; Helin, K.; Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733-750.
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen- Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494.e15.
- Chung, J.; Karkhanis, V.; Tae, S.; Yan, F.; Smith, P.; Ayers, L.W.; Agostinelli, C.; Pileri, S.; Denis, G.V.; Baiocchi, R.A.; et al. Protein arginine methyltransferase 5 (PRMT5) inhibition induces lymphoma cell death through reactivation of the retinoblastoma tumor suppressor pathway and polycomb repressor complex 2 (PRC2) Silencing. J. Biol. Chem. 2013, 288, 35534–35547.
- Poole, C.J.; van Riggelen, J.; MYC—master regulator of the cancer epigenome and transcriptome. Genes 2017, 8, 142.
- Fujita, N.; Jaye, D.L.; Geigerman, C.; Akyildiz, A.; Mooney, M.R.; Boss, J.M.; Wade, P.A.; MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell 2004, 119, 75-86.
- Lemercier, C.; Brocard, M.P.; Puvion-Dutilleul, F.; Kao, H.Y.; Albagli, O.; Khochbin, S.; Class II histone deacetylases are directly recruited by BCL6 transcriptional repressor. J. Biol. Chem. 2002, 277, 22045-22052.
- Gearhart, M.D.; Corcoran, C.M.; Wamstad, J.A.; Bardwell, V.J.; Polycomb Group and SCF Ubiquitin Ligases Are Found in a Novel BCOR Complex That Is Recruited to BCL6 Targets. Mol. Cell. Biol. 2006, 26, 6880-6889.
- Pasqualucci, L.; Dominguez-Sola, D.; Chiarenza, A.; Fabbri, G.; Grunn, A.; Trifonov, V.; Kasper, L.H.; Lerach, S.; Tang, H.; Ma, J.; et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature 2011, 471, 189-195.
- Berdasco, M.; Esteller, M.; Clinical epigenetics: Seizing opportunities for translation. Nat. Rev. Genet. 2019, 20, 109-127.
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Götze, K.S.; A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenetics 2016, 8, 1-11.
More
Information
Subjects:
Hematology; Oncology; Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.6K
Revisions:
2 times
(View History)
Update Date:
13 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No