Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Bahauddeen M. Alrfaei | + 2402 word(s) | 2402 | 2022-01-05 09:02:36 | | | |
| 2 | Conner Chen | Meta information modification | 2402 | 2022-01-13 06:54:51 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Alrfaei, B. MTOR Inhibitor Resistance in Medulloblastoma. Encyclopedia. Available online: https://encyclopedia.pub/entry/18149 (accessed on 04 July 2026).
Alrfaei B. MTOR Inhibitor Resistance in Medulloblastoma. Encyclopedia. Available at: https://encyclopedia.pub/entry/18149. Accessed July 04, 2026.
Alrfaei, Bahauddeen. "MTOR Inhibitor Resistance in Medulloblastoma" Encyclopedia, https://encyclopedia.pub/entry/18149 (accessed July 04, 2026).
Alrfaei, B. (2022, January 12). MTOR Inhibitor Resistance in Medulloblastoma. In Encyclopedia. https://encyclopedia.pub/entry/18149
Alrfaei, Bahauddeen. "MTOR Inhibitor Resistance in Medulloblastoma." Encyclopedia. Web. 12 January, 2022.
Copy Citation
Medulloblastoma is a common fatal pediatric brain tumor. The mechanistic target of rapamycin (mTOR) is one of the major pathways that have been activated during medulloblastoma development. It is a master regulator for signaling pathways that organize organismal development and homeostasis, because of its involvement in protein and lipid synthesis besides controlling the cell cycle and the cellular metabolism. mTOR inhibitors are a class of drugs that suppress the mTOR. In the clinic, they are primarily used as immunosuppressants and for the treatment of multiple cancers. Three generations of mTOR-targeted therapy have been developed to date. Resistance has been observed against mTOR inhibitor-targeted therapy in medulloblastoma.
mTOR
medulloblastoma
targeted therapy
resistance
1. mTOR Molecular Pathway
The mechanistic target of rapamycin (mTOR) is a serin-threonine kinase that belongs to the phosphatidylinositol 3-kinase-related kinases [1]. It compromises part of two distinct protein complexes, mTORC1 and mTORC2 [2]. mTORC1 consists of a regulatory protein associated with mTOR with Raptor, mLST8, PRAS40, and DEPTOR. mTORC2 is characterized by Rictor, the rapamycin-insensitive companion of mTOR, in addition to DEPTOR, mLST8, mSin1, and Protor1/2 [1]. Secondary to external or internal stimuli, both complexes interact in multiple signaling pathways, such as the phosphoinositide 3-kinases (PI3K)/Akt/mTOR signaling cascade and mitogen-activated protein kinase (MAPK) signaling cascade [2]. mTOR has a central role in the regulation of cellular metabolism, including anabolic processes, cellular growth, proliferation, and autophagy [3]. mTOR is also involved in many embryonal processes such as T cell differentiation with neurogenesis, synaptic formation, and neural stem cell regulation [4][5]. The involvement of mTOR in cell differentiation of T helpers: Th1, Th2, and Th17 have been investigated. Inhibition of Rheb (Ras homolog) reduces Th1 differentiation. Similarly, Raptor deletion reduces Th2 differentiation. Raptor deficiency resulted in a significant decrease of Th17+ CD27− differentiation to Th17 CD27, which is responsible for producing IFN-ɣ. The deletion of Rictor showed a decrease in both Th1 and Th2 cell differentiation through the downregulation of the AKT and PKC pathways, respectively [4][6].
mTORC1 and mTORC2 expression are important in neural stem cell (NSC) differentiation and prenatal neurogenesis. However, embryonic stem cells express a low level of mTORC1/p70S6K to maintain their undifferentiated state through the upregulation of tuberous sclerosis complex 1/2 (TSC1/2) and DEPTOR, which are mTORC1 inhibitors. The activation of Raptor is specific for NSC differentiation and migration in corticogenesis. In addition, the inhibition of TSC1 and the activation of Rheb promote cortical progenitor cell differentiation and migration. The mTORC1 critical role in maintaining an appropriate cell size and number is compromised when Raptor is deleted resulting in microcephaly. The high expression of mTORC2 is vital for cytoskeletal function and for the interaction with AKT [5]. In cortical neurons, the mTOR signaling is enhanced by dendritic branching when PTEN is deleted. Similarly, the dendritic branching is enhanced by TSC1 deletion and Rheb activation. Although the Dendritic branching is reduced by mTOR inhibition, the reduction is rescued by mTOR resistant mutation [7]. The activation of mTORC1 in the olfactory granule neurons increases dendritic complexity. mTORC1 specific inhibition does not reduce dendrite complexity, but non-specific mTOR inhibition affects dendrite branching [5]. The deletion of TSC1/2 reduces synaptic formation and maintenance [8]. In mice, heterozygous TSC2 develops a dense dendritic spine at the first month of age due to pruning defects. Rapamycin treatment enhances spine elimination in TSC2 heterozygous mice by inducing autophagy. mTORC1 activity is crucial for synapse formation and maintenance. The dysregulation of its activity results in epilepsy and autism spectrum disorder [5].
Dysregulation of the more complexes (mTORC1 and mTORC2) are involved in many pathophysiological processes, including neurodegeneration, metabolic disorders, and tumorigenesis (Figure 1) [9]. The hyperactivation of mTOR underpins the mechanisms of multiple cancers. This overactivation is mainly secondary to the genetic mutations, activation of upstream molecules, or loss of inhibitory proteins such as PTEN [5][9]. This hyperactivation suggests mTOR resistance to treatment.
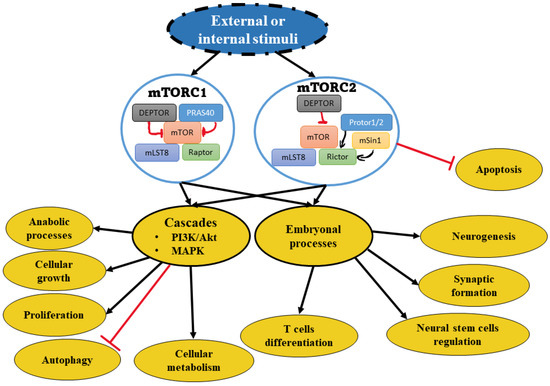
Figure 1. Schematic view of mTOR complexes and relevant pathways. The depiction shows mTOR complexes that induce downstream pathway activation. The activation involves either or both cascade cellular activities and/or embryonal processes under development.
2. mTOR Involvement in Medulloblastoma
In medulloblastoma, mTOR has a fundamental role in tumor development through many pathways. The Sonic hedgehog (SHH) pathway is critical for cell fate determination and embryonic neuronal development, and it is implicated in multiple cancers, including the SHH subgroup in medulloblastoma. SHH is a secreted protein which when bound to a cellular membrane protein PTCH1, releases its inhibitor on the smoothed homolog (Smo). The Smo protein transduces Gli protein which is a transcription factor, leading to downstream gene expression adjustments [10]. mTORC1 affects this process through a translational effect on Smo, resulting in the hyperactivation and proliferation of granule neuron precursor cells [11]. The mTOR pathway is implicit in the development of medulloblastoma stem cells (cancer stem cells), resulting in treatment failure [12]. It is known that cancer stem cells are resistant to conventional therapy [13]. The crosstalk between mTOR and Hedgehog (HH) pathways has been reported to stimulate medulloblastoma progression. This crosstalk promotes mRNA translation by stimulating the expression of eukaryotic translation initiation factor 4E (eIF4E) and inhibiting eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1), leading to medulloblastoma tumor progression [11]. The factor eIF4E is present downstream of PI3K. In inhibition of both (4EBP1 and eIF4E) proteins may induce synergistic effect as reported in breast cancer cell lines [14]. Similarly, dual targeting of HH and PI3K/mTOR pathways in a medulloblastoma mouse model, inhibited tumor growth and extended survival of tumor-bearing animals [15]. In addition, SHH-driven medulloblastoma is inhibited by targeting the mTOR pathway [11]. Taken together, these findings suggest that dual targeting mTOR signaling pathway in addition to other pathways is a promising therapeutic approach to overcome resistance in treating medulloblastoma patients.
Most of the medulloblastoma molecular subgroups harbor genetic and epigenetic alterations that activate the PI3K/Akt/mTOR pathway to fuel cancer progression [16][17]. For instance, various genetic alterations in the PIK3CA gene have been detected in both group 4 and the WNT subgroup. The PI3K/Akt/mTOR pathway activation has also been reported within the MYC-driven group 3 medulloblastoma [18]. Interestingly, the combination of PIK inhibitors with histone deacetylase inhibitors (HDACI), reduced the tumor growth of MCY-driven medulloblastoma in vivo, providing a novel therapeutic approach to treating the most aggressive form of medulloblastoma. Recently, a combination of siRNA and small molecule inhibitors to target the MYC and mTOR pathways in MYC-driven medulloblastoma cells resulted in reducing cell growth and survival in vitro and prolonged survival of MYC-driven medulloblastoma xenografts [19]. In addition to the contribution of genetic and epigenetic activation of The PI3K/Akt/mTOR pathway in medulloblastoma progression, aberrant constitutive activation of several growth factor receptors acting upstream of the PI3K/Akt/mTOR pathway has also been linked with the development and progression of some of the medulloblastoma subgroups. For instance, medulloblastoma group 4, which is considered the largest and the most diverse group, harbors multiple molecular alterations that constitutively activate several growth factor receptors, including the insulin-like growth factor (IGF), platelet-derived growth factor (PDGF), epidermal endothelial growth factor (EGFR) and vascular endothelial growth factor (VEGF) among others [20][21][22]. It has been suggested that targeting cell membrane receptors that are highly expressed on medulloblastoma cells could be one of the effective therapeutic approaches to medulloblastoma treatment. In that direction, Snuderl et.al, reported that placental growth factor (P1GF) and its receptor neuropilin1 (Nrp1) are highly expressed on the majority of medulloblastoma cells and their expressions are associated with poor survival and metastasis in patients [23]. Blocking P1GF/Nrp1 in medulloblastoma animal models resulted in disease regression, reduction of metastasis and prolonged survival of treated animals [23]. All these findings suggest that the mTOR signaling is a key regulator of medulloblastoma development and progression and is considered a therapeutic target for treating medulloblastoma patients in combination with other current therapies.
3. mTOR-Targeted Therapy in Medulloblastoma
Multiple drugs have been reported to target mTOR through different binding sites or mechanisms. Rapamycin inhibits mTORC1 through binding to FK506 Binding Protein 12 (FKBP12). This forms a complex which binds to FKB12-Rapamycin Binding (FRB), an active site at mTOR. The complex triggers a structural modification that causes the fragmentation of Raptor and mTOR binding [24]. Similarly, rapamycin analogs or first-generation mTOR inhibitors such as temsirolimus and everolimus, are targeting mTORC1 and causing disintegration through allosteric inhibition with altered pharmaceutical properties (Figure 2) [25][26].
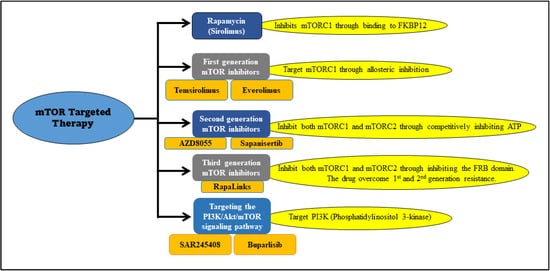
Figure 2. mTOR inhibitors and their targets. The depiction illustrates specific inhibitors and their targets used for mTOR-targeted therapy.
Temsirolimus is a prodrug of sirolimus [27]. Sirolimus (rapamycin) was initially approved for immunosuppression, followed by studies investigating other potential roles, including its antineoplastic effect. Sirolimus has completed a phase I clinical trial for pediatric solid tumors ((NCT01331135) such as ewing sarcoma, osteosarcoma, glioblastoma multiforme, ependymoma, and medulloblastoma [28]. The study goals were to examine the tolerated doses, the safety profile of sirolimus, understand the correlation with the mTOR effector pS6 kinase, and the risk of infections. In total, 18 patients were treated with escalating doses of sirolimus in combination with celecoxib, etoposide, and cyclophosphamide. Only two medulloblastoma patients were included in this study. The study found that the drugs were well tolerated, with a decrease in the lymphocyte count CD4 as a side effect, and the pS6 levels were undetectable in all the sirolimus dosing regimens [28].
Temsirolimus (CCI-779) is formed through the esterification of rapamycin, making it more water-soluble and easily administered through the intravenous route. It is converted to rapamycin through carboxylesterases, specifically by CYP3A4 and CYP3A5 [27]. Temsirolimus is a kinase inhibitor which has been studied in multiple cancers such as renal cancer, ovarian cancer, lymphoma and neuroblastoma [25]. It completed a phase III clinical trial for refractory renal cell carcinoma with significant increase in survival rate compared to the interferon-alpha version of the drug. It has been approved by FDA for renal cell carcinoma [29]. Temsirolimus has been evaluated in medulloblastoma within three phases I clinical trials which were evaluating multiple pediatric solid tumors. The first clinical trial included 18 patients of refractory solid tumors who were treated with temsirolimus, and it was well tolerated, except for nausea as the major dose-limiting toxicity [30]. In this trial, only two medulloblastoma patients were recruited. Similarly, to everolimus, the phosphorylations of AKT, pS6, and 4EBP1 were decreased; however, it was not related to dose or clinical response [30]. The second study enrolled 71 pediatric patients with relapsed tumors. This study has only two medulloblastoma patients’ recruited. The study participants were treated with an increasing dose of intravenous (IV) temsirolimus, oral irinotecan and temozolomide. These treatments were well tolerated, except for hyperlipidemia as dose-limiting toxicity and few adverse events [31]. The third phase I clinical trial was to investigate temsirolimus combined with perifosine in pediatric solid tumors (Table 1) [32]. Perifosine is an antitumor alkyl phospholipid (APL) that targets AKT [33]. The study goals were to determine the toxicity, the dosing amount, the pharmacokinetics of the medications, and the effects of the combination of the AKT inhibitor with the mTOR inhibitor. In total, 22 patients were recruited for the combination therapy, which was safe and well-tolerated. There was a linear correlation between the perifosine dose and response. However, significant plasma level variability was documented in patients which prevents proper assessment of side effects [32]. A preclinical xenograft study was conducted using a medulloblastoma model implanted PNET/MB cell line (DAOY) that is rapamycin-sensitive. The model was treated with rapamycin, temsirolimus, and cisplatin, which had an additive cytotoxicity effect. The single-agent treatment with temsirolimus decreased growth significantly after one week of treatment [34].
Table 1. mTOR inhibitors clinical trials in medulloblastoma (MD). The table summarizes completed and current clinical trials that used mTOR inhibitors in the treatment of medulloblastoma.
| Drug | Target | Patients Groups | Medulloblastoma Cases/Total Tumor Cases |
Phase | Status/ Result | The National Clinical Trial Number |
|---|---|---|---|---|---|---|
| Sirolimus in combination with metronomic therapy |
mTOR | Children with recurrent or refractory solid and brain tumors |
2 / 18 | I | Complete/ well tolerated |
NCT01331135 |
| Everolimus | mTOR | Pediatric patients with refractory solid tumors | 3 / 41 | I | Complete/ well tolerated |
NCT00187174 |
| Temsirolimus | mTOR | Pediatric patients with recurrent/refractory solid tumors | 2 / 71 | I | Complete/ did not meet efficacy |
NCT00106353. |
| Temsirolimus in combination with irinotecan and temozolomide | mTOR | Children, adolescents, and young adults with relapsed or refractory solid tumors | 2 / 72 | I | Complete/ tolerated dose | NCT01141244 |
| Temsirolimus with perifosine | mTOR AKT |
Recurrent pediatric solid tumors |
2 / 23 | I | Complete/ tolerable toxicity | NCT01049841 |
| Vismodegib in combination with temozolomide versus temozolomide alone | Smo mTOR |
Patients with medulloblastomas with an activation of the Sonic hedgehog pathway | 24 / 24 | I II |
Terminated/ unclear |
NCT01601184 |
Everolimus is an analog with higher bioavailability than sirolimus [35]. It has been investigated in clinical trials for various tumors, such as non-small cell carcinoma, mantle cell lymphoma, Rhabdomyosarcoma Sarcoma, and medulloblastoma (NCT00187174) [35]. Everolimus is FDA approved for neuroendocrine tumors of pancreatic origin (PNET) [35]. A clinical trial phase I to assess everolimus was reported for medulloblastoma and other pediatric tumors. The maximum tolerated dose of everolimus was evaluated through 41 participants. The study demonstrated inhibition of the mTOR pathway in peripheral blood mononuclear cell including downstream molecules p70-S6 kinase, and AKT. These molecules usually get activated by mTORC2 stimulation cascade [36]. The treatment was well tolerated in children with solid tumors. Minimal pS6 kinase activity and a decrease in AKT phosphorylation were observed after therapy. However, in the adult population increase in phosphorylation and higher expression of mTORC2 were observed post-treatment. A possible theory for the treatment failure was the development of resistance against first-generation mTOR inhibitor [36][37].
The second-generation mTOR inhibitors (Figure 2) affect both mTORC1 and mTORC2 through competitively binding to the ATP sites. They also have a PI3K inhibitory effect [25]. Many inhibitors are being tested for various cancers such as GSK2126458 and gedatolisib, but not yet tested on medulloblastoma [20]. AZD8055 is a potent second-generation inhibitor with mTORC1, mTORC2, and PI3K inhibitory effects (Table 1) [38]. It completed a phase I clinical trial in patients with solid tumors, including breast, lung, and pancreatic cancers, but is still in a preclinical (animal studies) phase regarding medulloblastoma [39]. There is a study used medulloblastoma xenograft (BT-50) cells with AZD8055 treatment resulted in a stable disease status [40]. Similarly, another drug called sapanisertib (MLN0128) was tested on medulloblastoma (BT-28 cells) xenograft, which induced disease stabilization but not regression [41].
The third-generation mTOR inhibitors, also called RapaLinks, were developed to overcome resistant mutations of the first and second generations in the FKBP12-rapamycin binding (FRB) or kinase domain mutants through an avidity-based approach [42]. This generation does not inhibit rapamycin binding to FKBP12 or the FRB domain of mTOR. It delivers MLN0128 to inhibit the ATP site of the mTORC1 complex. The third generation showed promising results after they induced initial regression in glioblastoma [43]. Furthermore, no clinical studies have been conducted on the third generation against medulloblastoma malignancy.
References
- Yang, H.; Rudge, D.G.; Koos, J.D.; Vaidialingam, B.; Yang, H.J.; Pavletich, N.P. mTOR Kinase Structure, Mechanism and Regulation by the Rapamycin-Binding Domain. Nature 2013, 497, 217–223.
- Jhanwar-Uniyal, M.; Wainwright, J.V.; Mohan, A.L.; Tobias, M.E.; Murali, R.; Gandhi, C.D.; Schmidt, M.H. Diverse Signaling Mechanisms of mTOR Complexes: MTORC1 and MTORC2 in Forming a Formidable Relationship. Adv. Biol. Regul. 2019, 72, 51–62.
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976.
- Yan, J.; Wang, R.; Horng, T. MTOR Is Key to T Cell Transdifferentiation. Cell Metab. 2019, 29, 241–242.
- LiCausi, F.; Hartman, N.W. Role of mTOR Complexes in Neurogenesis. Int. J. Mol. Sci. 2018, 19, 1544.
- Huang, H.; Long, L.; Zhou, P.; Chapman, N.M.; Chi, H. MTOR Signaling at the Crossroads of Environmental Signals and T-Cell Fate Decisions. Immunol. Rev. 2020, 295, 15–38.
- Jaworski, J.; Spangler, S.; Seeburg, D.P.; Hoogenraad, C.C.; Sheng, M. Control of Dendritic Arborization by the Phosphoinositide-3′-kinase-akt-mammalian Target of Rapamycin Pathway. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 11300–11312.
- Bateup, H.S.; Takasaki, K.T.; Saulnier, J.L.; Denefrio, C.L.; Sabatini, B.L. Loss of tsc1 In Vivo Impairs Hippocampal Mglur-ltd and Increases Excitatory Synaptic Function. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 8862–8869.
- Murugan, A.K. MTOR: Role in Cancer, Metastasis and Drug Resistance. Semin. Cancer Biol. 2019, 59, 92–111.
- Li, X.; Li, Y.; Li, S.; Li, H.; Yang, C.; Lin, J. The Role of Shh Signalling Pathway in Central Nervous System Development and Related Diseases. Cell Biochem. Funct. 2021, 39, 180–189.
- Wu, C.-C.; Hou, S.; Orr, B.A.; Kuo, B.R.; Youn, Y.H.; Ong, T.; Roth, F.; Eberhart, C.G.; Robinson, G.W.; Solecki, D.J.; et al. MTORC1-Mediated Inhibition of 4EBP1 Is Essential for Hedgehog Signaling-Driven Translation and Medulloblastoma. Dev. Cell 2017, 43, 673–688.
- Kumar, V.; Kumar, V.; McGuire, T.; Coulter, D.W.; Sharp, J.G.; Mahato, R.I. Challenges and Recent Advances in Medulloblastoma Therapy. Trends Pharmacol. Sci. 2017, 38, 1061–1084.
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma Stem Cells Promote Radioresistance by Preferential Activation of the DNA Damage Response. Nature 2006, 444, 756–760.
- Lineham, E.; Tizzard, G.J.; Coles, S.J.; Spencer, J.; Morley, S.J. Synergistic effects of inhibiting the mnk-eif4e and pi3k/akt/ mTOR pathways on cell migration in mda-mb-231 cells. Oncotarget 2018, 9, 14148–14159.
- Chaturvedi, N.K.; Kling, M.J.; Coulter, D.W.; McGuire, T.R.; Ray, S.; Kesherwani, V.; Joshi, S.S.; Sharp, J.G. Improved therapy for medulloblastoma: Targeting hedgehog and pi3k-mTOR signaling pathways in combination with chemotherapy. Oncotarget 2018, 9, 16619.
- Dimitrova, V.; Arcaro, A. Targeting the PI3K/AKT/MTOR Signaling Pathway in Medulloblastoma. Curr. Mol. Med. 2015, 15, 82–93.
- Robinson, G.; Parker, M.; Kranenburg, T.A.; Lu, C.; Chen, X.; Ding, L.; Phoenix, T.N.; Hedlund, E.; Wei, L.; Zhu, X.; et al. Novel Mutations Target Distinct Subgroups of Medulloblastoma. Nature 2012, 488, 43–48.
- Pei, Y.; Liu, K.-W.; Wang, J.; Garancher, A.; Tao, R.; Esparza, L.A.; Maier, D.L.; Udaka, Y.T.; Murad, N.; Morrissy, S.; et al. HDAC and PI3K Antagonists Cooperate to Inhibit Growth of MYC-Driven Medulloblastoma. Cancer Cell 2016, 29, 311–323.
- Chaturvedi, N.K.; Kling, M.J.; Griggs, C.N.; Kesherwani, V.; Shukla, M.; McIntyre, E.M.; Ray, S.; Liu, Y.; McGuire, T.R.; Sharp, J.G. A novel Combination Approach Targeting an Enhanced Protein Synthesis Pathway in Myc-driven (group 3) Medulloblastoma. Mol. Cancer Ther. 2020, 19, 1351–1362.
- Aldaregia, J.; Odriozola, A.; Matheu, A.; Garcia, I. Targeting MTOR as a Therapeutic Approach in Medulloblastoma. Int. J. Mol. Sci. 2018, 19, 1838.
- Cavalli, F.M.G.; Remke, M.; Rampasek, L.; Peacock, J.; Shih, D.J.H.; Luu, B.; Garzia, L.; Torchia, J.; Nor, C.; Morrissy, A.S.; et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell 2017, 31, 737–754.
- Paul, R.; Bapat, P.; Deogharkar, A.; Kazi, S.; Singh, S.K.V.; Gupta, T.; Jalali, R.; Sridhar, E.; Moiyadi, A.; Shetty, P.; et al. MiR-592 Activates the MTOR Kinase, ERK1/ERK2 Kinase Signaling and Imparts Neuronal Differentiation Signature Characteristic of Group 4 Medulloblastoma. Hum. Mol. Genet. 2021, 30, 2416–2428.
- Snuderl, M.; Batista, A.; Kirkpatrick, N.D.; de Almodovar, C.R.; Riedemann, L.; Walsh, E.C.; Anolik, R.; Huang, Y.; Martin, J.D.; Kamoun, W. Targeting Placental Growth Factor/neuropilin 1 Pathway Inhibits Growth and Spread of Medulloblastoma. Cell 2013, 152, 1065–1076.
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a Protein Target of the FKBP12-Rapamycin Complex in Mammalian Cells. J. Biol. Chem. 1995, 270, 815–822.
- Chen, Y.; Zhou, X. Research Progress of mTOR Inhibitors. Eur. J. Med. Chem. 2020, 208, 112820.
- Meng, L.; Zheng, X.S. Toward Rapamycin Analog (Rapalog)-Based Precision Cancer Therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169.
- Mizuno, T.; Fukuda, T.; Christians, U.; Perentesis, J.P.; Fouladi, M.; Vinks, A.A. Population Pharmacokinetics of Temsirolimus and Sirolimus in Children with Recurrent Solid Tumours: A Report from the Children’s Oncology Group. Br. J. Clin. Pharmacol. 2017, 83, 1097.
- Qayed, M.; Cash, T.; Tighiouart, M.; MacDonald, T.J.; Goldsmith, K.C.; Tanos, R.; Kean, L.; Watkins, B.; Suessmuth, Y.; Wetmore, C.; et al. A phase i study of sirolimus in combination with metronomic therapy (choanome) in children with recurrent or refractory solid and brain tumors. Pediatr. Blood Cancer 2020, 67, e28134.
- Hudes, G.; Carducci, M.; Tomczak, P.; Dutcher, J.; Figlin, R.; Kapoor, A.; Staroslawska, E.; Sosman, J.; McDermott, D.; Bodrogi, I.; et al. Temsirolimus, Interferon Alfa, or Both for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2009, 356, 2271–2281.
- Spunt, S.L.; Grupp, S.A.; Vik, T.A.; Santana, V.M.; Greenblatt, D.J.; Clancy, J.; Berkenblit, A.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; et al. Phase I Study of Temsirolimus in Pediatric Patients With Recurrent/Refractory Solid Tumors. J. Clin. Oncol. 2011, 29, 2933.
- Bagatell, R.; Norris, R.; Ingle, A.; Ahern, C.; Voss, S.; Fox, E.; Little, A.; Weigel, B.; Adamson, P.; Blaney, S. Phase 1 Trial of Temsirolimus in Combination with Irinotecan and Temozolomide in Children, Adolescents and Young Adults with Relapsed or Refractory Solid Tumors: A Children’s Oncology Group Study. Pediatr. Blood Cancer 2014, 61, 833–839.
- Becher, O.J.; Gilheeney, S.W.; Khakoo, Y.; Lyden, D.C.; Haque, S.; De Braganca, K.C.; Kolesar, J.M.; Huse, J.T.; Modak, S.; Wexler, L.H.; et al. A Phase I Study of Perifosine with Temsirolimus for Recurrent Pediatric Solid Tumors. Pediatr. Blood Cancer 2017, 64, 1–9.
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a Novel Akt Inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110.
- Geoerger, B.; Kerr, K.; Tang, C.-B.; Fung, K.-M.; Powell, B.; Sutton, L.N.; Phillips, P.C.; Janss, A.J. Antitumor Activity of the Rapamycin Analog CCI-779 in Human Primitive Neuroectodermal Tumor/Medulloblastoma Models as Single Agent and in Combination Chemotherapy. Cancer Res. 2001, 61, 1527–1532.
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One Drug, Many Effects. Cell Metab. 2014, 19, 373–379.
- Fouladi, M.; Laningham, F.; Wu, J.; O’Shaughnessy, M.A.; Molina, K.; Broniscer, A.; Spunt, S.L.; Luckett, I.; Stewart, C.F.; Houghton, P.J.; et al. Phase I Study of Everolimus in Pediatric Patients With Refractory Solid Tumors. J. Clin. Oncol. 2016, 25, 4806–4812.
- Dancey, J.E. Therapeutic Targets: MTOR and Related Pathways. Cancer Biol. Ther. 2006, 5, 1065–1073.
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 Is a Potent, Selective, and Orally Bioavailable ATP-Competitive Mammalian Target of Rapamycin Kinase Inhibitor with In Vitro and In Vivo Antitumor Activity. Cancer Res. 2010, 70, 288–298.
- Asahina, H.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Tamura, Y.; Honda, K.; Seki, Y.; Tanabe, Y.; Shimada, H.; Shi, X.; et al. Safety and Tolerability of AZD8055 in Japanese Patients with Advanced Solid Tumors; A Dose-Finding Phase i Study. Invest. New Drugs 2013, 31, 677–684.
- Houghton, P.J.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Morton, C.L.; Keir, S.T.; Reynolds, C.P.; Kang, M.H.; Phelps, D.; et al. Initial Testing (Stage 1) of the mTOR Kinase Inhibitor AZD8055 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2012, 58, 191–199.
- Kang, M.H.; Reynolds, C.P.; Maris, J.M.; Gorlick, R.; Kolb, E.A.; Lock, R.; Carol, H.; Keir, S.T.; Wu, J.; Lyalin, D.; et al. Initial Testing (Stage 1) of the Investigational MTOR Kinase Inhibitor MLN0128 by the Pediatric Preclinical Testing Program. Pediatr. Blood Cancer 2014, 61, 1486–1489.
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming MTOR Resistance Mutations with a New Generation MTOR Inhibitor. Nature 2016, 534, 272–276.
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.-Q.; Cavanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to MTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435.
More
Information
Subjects:
Oncology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
738
Revisions:
2 times
(View History)
Update Date:
13 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No