Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yaping Liu | + 3871 word(s) | 3871 | 2022-01-05 08:47:18 | | | |
| 2 | Camila Xu | Meta information modification | 3871 | 2022-01-12 02:06:12 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Liu, Y. Protein Quality Control System. Encyclopedia. Available online: https://encyclopedia.pub/entry/18094 (accessed on 25 July 2026).
Liu Y. Protein Quality Control System. Encyclopedia. Available at: https://encyclopedia.pub/entry/18094. Accessed July 25, 2026.
Liu, Yaping. "Protein Quality Control System" Encyclopedia, https://encyclopedia.pub/entry/18094 (accessed July 25, 2026).
Liu, Y. (2022, January 12). Protein Quality Control System. In Encyclopedia. https://encyclopedia.pub/entry/18094
Liu, Yaping. "Protein Quality Control System." Encyclopedia. Web. 12 January, 2022.
Copy Citation
As a collection of pathways that regulates proteins’ life cycles including their synthesis, folding, assembly, degradation and reversal, the protein quality control system mainly consists of the ERS, ALS and UPS. It plays an important role in maintaining normal cell metabolism and avoiding protein dysfunction, especially in the physiological and pathological processes of AD.
Alzheimer’s disease
protein quality control
endoplasmic reticulum stress
1. Introduction
Alzheimer’s disease (AD) is an aging-related neurodegenerative disorder accompanied by memory loss, cognitive impairment, synaptic damage and behavioral changes [1]. As one of the most principal forms of dementia, it accounts for 70% of patients with dementia [2][3][4][5]. Currently, it is estimated that approximately 47 million people suffer from dementia worldwide [6] and with the aggravation of global population aging, the number of people with dementia cases will escalate to 74.7 million by 2030 and 131.5 million by 2050 [7][8]. Presently, growing evidence indicates that the main clinical manifestations of AD mostly occur after 65 years of age [9], including aphasia, apraxia, agnosia, an incapacity for discernment, and changes in personality and behavior and culminating in an individual’s death [10][11][12].
However, it should be noted that as the most common chronic neurological disease in the elderly, the challenge of AD treatment at present is the absence of an effective combination of sensitive biomarkers for early diagnosis. Some studies have shown that the pathological biomarkers of AD mainly include extracellular deposition of senile plaques (SPs) composed of amyloid-β protein (Aβ) and intracellular accumulation of neurofibrillary tangles (NFTs) consisting of hyperphosphorylated Tau (p-Tau), as well as neuronal loss in different brain regions. In fact, patients have shown a sustained disease progression for at least 10 to 15 years before these biomarkers are detected [13]. Thus, the challenge of early diagnosis of AD is far from being solved. Furthermore, the current approaches to treating AD are still severely lagging. In spite of drugs aimed at relieving the symptoms of AD, patients have been widely studied for a long time, which has largely been ineffective or inconclusive ultimately due to a variety of reasons [14]. For example, the drug that targets Aβ has been proved to provide symptomatic relief for only the initial 1–2 years, but is incapable of preventing or delaying the progression of the AD pathology fundamentally [8][15]. With the changes in dietary structure and the increase of life expectancy, AD has become the most devastating neurodegenerative disorder characterized by a high morbidity rate and mortality [14][16]. Simultaneously, it also brings a heavy economic burden [2], and even has the potential to evolve into a global public health concern if left unchecked [10][17][18][19][20].
2. The Protein Quality Control System
As a collection of pathways that regulates proteins’ life cycles including their synthesis, folding, assembly, degradation and reversal, the protein quality control system mainly consists of the ERS, ALS and UPS [21]. It plays an important role in maintaining normal cell metabolism and avoiding protein dysfunction, especially in the physiological and pathological processes of AD [22]. Various evidence shows that when abnormal protein accumulates, as an adaptive response of the ERS, the unfolded protein response (UPR) is provoked to produce normal proteins by up-regulating the expression of molecular chaperones, while reducing the accumulation of misfolded proteins via decelerating the synthesis of total proteins [23]. In addition, the adaptive and protective interactions between the ERS and UPS can help cells to clear toxic protein aggregation, reduce the imbalance in the endoplasmic reticulum lumen, and restore cell homeostasis [1]. However, the continuous imbalance of endoplasmic reticulum homeostasis, as well as autophagy dysfunctions could mediate the gradual transformation from the ERS to autophagy, that is, from an adaptive and protective state to a persistent and destructive condition [23]. For example, if the ERS fails to refold the abnormal proteins for some reason, the molecular chaperones would deliver the abnormal proteins to the autophagy–lysosome system or ubiquitin–proteasome system, where the abnormal proteins can be effectively degraded [22]. During the progression of AD, due to the abnormal expression and impaired function of key components of these pathways, as well as deficits in protein homeostasis, the abnormal protein would be encapsulated in the proteasome and lysosome to form the endosome and be degraded unsuccessfully, which finally induces dysregulation of proteostasis and pathological damage (Figure 1).
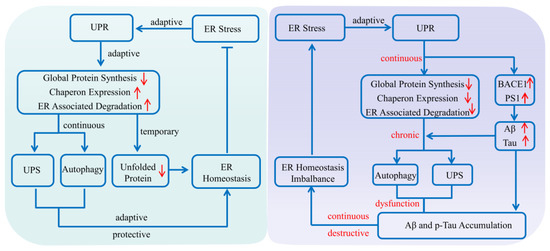
Figure 1. Protein quality control system affecting Alzheimer’s disease. The endoplasmic reticulum system, autophagy–lysosome system and ubiquitin–proteasome system are the three main regulatory pathways in maintaining normal cell metabolism and avoiding protein dysfunction. Once abnormal protein accumulates in the brain, the unfolded protein response is initially provoked to produce normal proteins by upregulating the expression of molecular chaperones, while reducing the accumulation of misfolded proteins via inhibiting the synthesis of total proteins. If the endoplasmic reticulum system fails to refold the abnormal protein for some reason, the molecular chaperones will deliver the abnormal proteins to the autophagy–lysosome system or the ubiquitin–proteasome system, where the abnormal proteins can be effectively degraded. While during AD progression, all the abnormal expression and impaired function of key components of these pathways, as well as defects in the proteins’ interplay, could induce dysregulation of proteostasis and contribute to AD pathogenesis. (Aβ: amyloid-β protein; BACE1: β-amyloid precursor protein cleaving enzyme 1; ER: endoplasmic reticulum; PS1: presenilin 1; p-Tau: hyperphosphorylated Tau; UPR: unfolded protein response; UPS: ubiquitin–proteasome system). The upward red arrow indicates up-regulation of expression, while the downward red arrow indicates downregulation of expression. The blue arrow indicates the activation of process, while the blue T arrow indicates the inhibition of process.
Increasing evidence shows that the deposition of abnormal proteins including Aβ and p-Tau in AD is associated with dysfunction of the protein quality control system in the brain [22]. In particular, during AD progression, abnormal expression and impaired function of key components, as well as defects in the proteins’ interplay could induce dysregulation of proteostasis and contribute to AD pathogenesis [22]. Therefore, it is of great importance to explore the interrelationship between the protein quality control system and the pathogenesis of AD, which will help us to further understand the mechanisms and consequences of proteostasis dysregulation in detail, and even provide a potential novel therapeutic strategy for AD.
2.1. Endoplasmic Reticulum System and AD
Endoplasmic reticulum is a principal eukaryotic organelle responsible for protein folding, modification and secretion, in addition to lipid synthesis and calcium storage [24][25]. Endoplasmic reticulum dysfunction caused by genetic mutations or environmental stimuli can lead to the accumulation of unfolded and misfolded proteins [26], which will trigger the UPR, subsequently resulting in a series of downstream reactions [27][28]. Current studies have shown that the UPR can not only reduce total protein synthesis by altering the intracellular transcription and translation processes [11], but also enhance the protein folding function and prevent the output of unfolded or misfolded proteins by up-regulating the molecular chaperones in the endoplasmic reticulum [26]. Moreover, it can also facilitate abnormal protein degradation via the ER-associated protein degradation (ERAD) pathway [11].
Under normal physiological conditions, there are three transmembrane ER-proximal sensors including protein kinase RNA-like ER kinase (PERK), activating transcription factor 6 (ATF6), and inositol-requiring protein 1 (IRE1) [29]. These ER sensors can form an inactive complex with the 78 kDa glucose-regulated protein (GRP78) [30], which acts as an ER chaperone that participates in the polypeptide translocation, thus being sequestered [29]. Nevertheless, under ERS conditions, the accumulating unfolded or misfolded proteins preferentially combine with GRP78 and activate the signaling pathways including the phosphorylation of PERK and IRE-1, as well as the translocation of ATF6 to the Golgi [25][30], which can regulate the expression of chaperones, decrease the accumulation of abnormal proteins, restore endoplasmic reticulum homeostasis and maintain cell functions [11][27]. Therefore, it can be concluded that in the early stage of the ERS, the UPR are mainly occurring to reduce the abnormal proteins in the ER by inhibiting the overall synthesis of proteins and clearing abnormal protein aggregations [28][31], so as to maintain the homeostasis of the ER [32]; However, if the ERS is persistent and unresolvable, the UPR will hyperactivate and even induce cell dysfunction and apoptosis [11][25][33].
Although AD has attracted much attention, its specific pathogenesis has not been fully elucidated [24][34][35][36][37]. Recently, a growing body of evidence demonstrates that the deposition of SPs and the formation of NFTs are not only salient features of AD, but linked to pathological ERS [38][39][40], highlighting the interrelationship of the ERS and AD [24][37][41][42]. In particular, the ERS is closely related to the production and accumulation of Aβ [17][43]. Under normal circumstances, the sequential cleavage of APP by α-secretase and γ-secretase occurs without the generation of Aβ. While in the pathological state of AD, the APP can be sequentially hydrolyzed by β-secretase and γ-secretase, and can then generate Aβ and induce toxicity cascade effects [44]. Liu et al. found that under the adaptive and protective ERS condition, the level of APP decreased in AD model cells induced by tunicamycin. In addition, the autopsy results of AD patients showed that the level of ER stress in the brain tissue had increased [45], indicating that the ERS might play an important role in AD [4][46][47] (Figure 2).
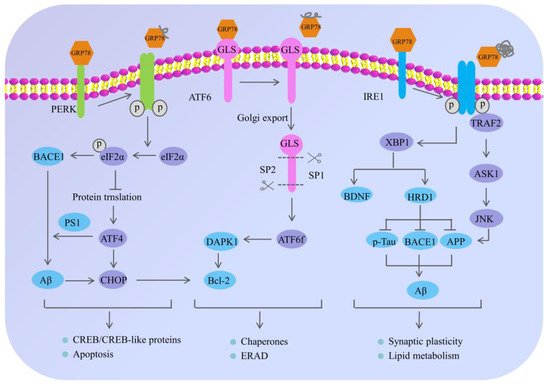
Figure 2. The mechanism of endoplasmic reticulum stress and its potential role in Alzheimer’s disease. Under normal physiological conditions, the ER sensors including PERK, ATF6 and IRE1 are inactivated through the interaction with 78 kDa glucose-regulated protein (GRP78); however, the misfolded proteins preferentially bind to GRP78, causing the dissociation of GRP78 from PERK, IRE1 and ATF6, eventually resulting in the phosphorylation of PERK and IRE-1, and the translocation of ATF6 to the Golgi. The activation of these signaling pathways regulates the expression of chaperones and decreases the accumulation of abnormal proteins, which can restore endoplasmic reticulum homeostasis. In neurons, under chronic ERS, the sustained activation of PERK leads to eIF2α phosphorylation, which not only influences the neuronal plasticity through protein synthesis inhibition, but also upregulates the expression of BACE1 and ATF4. Meanwhile, the BACE1 can be involved in the production of Aβ, and the ATF4 can further trigger cell death by upregulating the CHOP. Moreover, the adaptive activation of IRE1α leads to XBP1 splicing, which directly or indirectly participates in AD pathogenesis. On one hand, XBP1 can increase the degradation rate of key AD proteins—APP, BACE1 and p-Tau through inducing the E3 ubiquitin–ligase HRD1. On the other hand, the specific XBP1s’ splicing by IRE1 can also increase the generation of neurotrophic factor BDNF; however, the continuous activation of IRE1α leads to the preferential phosphorylation of TRAF2 and the inhibition of XBP1s splicing, which can further activate the downstream JNK signaling pathway and cause neuronal apoptosis. In addition, ATF6 is localized at the ER in physiological conditions and encodes a bZIP transcriptional factor in its cytosolic domain. While undergoing sustained ERS, ATF6 can translocate to the Golgi apparatus where it is processed by site 1 and 2 proteases releasing its cytosolic domain (ATF6f), and further controlling the upregulation of UPR target genes. The arrow indicates the activation of process, while the T arrow indicates the inhibition of process.
PERK, a type I transmembrane protein located in the ER, exerts serine/threonine kinase activity through its cytoplasmic domain [26]. During the early stage of the ERS, adaptive activation of PERK is a protective cellular mechanism [44]; however, persistent activation of PERK causes hyperphosphorylation of eIF2α at Ser51 [48], which can inhibit general translation initiation, lead to a reduction of critical memory proteins [17][49], further resulting in cognitive disorder and neurodegeneration [25][44][50]. Moreover, long-term sustained phosphorylation of eIF2α can also specifically up-regulate the expression of the β-amyloid precursor protein cleaving enzyme 1 (BACE1), which is a key enzyme responsible for initiating the generation of Aβ and promoting the formation of Aβ. Additionally, the hyperphosphorylated eIF2α can also activate the activating transcription factor 4 (ATF4). On one hand, ATF4, as a repressor of the cAMP response element binding protein (CREB)-dependent transcription, is responsible for long-term memory and synaptic plasticity [17][44], while the overexpression of ATF4 would severely impair memory function in AD. On the other hand, ATF4 mediates the abnormal processing of APP and promotes the excessive deposition of Aβ by upregulating the expression of presenilin 1 (PS1), which is an important cofactor for the production of Aβ. In addition, ATF4 can also act as a promoter of glycogen synthase kinase-3β (GSK-3β) expression to promote Tau hyperphosphorylation in AD patients [51]. Obviously, it suggested that the continuously activated PERK/eIF2α pathway may contribute to AD pathogenesis and cognitive impairments in many ways [44][52][53].
IRE1 is a transmembrane sensor kinase and an endoribonuclease that mediates both adaptive and proapoptotic pathways under ERS conditions [24]. The adaptive activation of IRE1α can lead to the splicing modification of XBP1 (a transcription factor of the leucine zipper family), which can up-regulate the expression of genes related to protein folding and promote the correct folding of proteins [31]. Moreover, XBP1 can increase not only the degradation rate of key AD proteins (APP, BACE1 and p-Tau) by inducing the E3 ubiquitin–ligase HRD1, but also the generation of neurotrophic factor BDNF [54]. Despite the initial activation of IRE1 signaling that may decrease the accumulation of abnormal proteins in AD, the continuous activation of IRE1 would mediate the phosphorylation of tumor necrosis factor receptor-associated factor 2 (TRAF2) and the inhibition of XBP1 splicing, which can trigger the c-Jun NH2-terminal kinase (JNK) signaling pathway and cause neuronal apoptosis [54][55]. To be specific, activated IRE1α on the ER membrane interacts with TRAF2, thus activating the following reactions: (1) it may recruit apoptosis signal-regulating kinase 1(ASK1), also known as MAP kinase, leading to activation of the mitochondria-dependent caspase apoptosis pathway [56][57]; (2) activated ASK1 further phosphorylates JNK and Bcl-2, eventually inducing apoptosis of nerve cells and aggravating nerve injury [58]; and (3) activated JNK can also in turn phosphorylate TRAF2, causing procaspase-12, which was originally bound to TRAF2, to be dissociated from the complex and cleaved after oligomerization to form active Caspase-12, leading to the occurrence of cell apoptosis [58]. Previous research showed that genetic ablation of the RNase domain of IRE1 in the nervous system significantly reduced the content of amyloid β oligomers, improved cognitive function and attenuated astrocyte activation [24]. At the molecular level, the deletion of IRE1 reduced the expression of APP in the cortical and hippocampal areas of AD mice [58].
Unlike PERK and IRE1 that belong to the endoplasmic reticulum type I transmembrane proteins family, ATF6 belongs to the endoplasmic reticulum type II transmembrane proteins family [55]. Once ERS occurs, GRP78 dissociates from ATF6 and is transported to the Golgi, where the active form of the ATF6 fragment with transcriptional activity is formed after the hydrolysis of protease site-1 and site-2. Then the ATF6 fragment is further transferred from the cytoplasm to the nucleus and regulating the expression of various genes related to ERS, such as GRP78 and protein disulfide isomerase, which promote protein folding and relieve ER pressure [23]. Meanwhile, the ATF6 cooperates with IRE1α to facilitate Xbp1-mediated transcription [59], and the ATF6 and XBP1 both activate PERK/eIF2α signaling, which suggests that there is an interaction among the three pathways of the URP. In addition, Du et al. found that ATF6 can reduce the expression of BACE1 by regulating the activity of the BACE1 promoter, thereby reducing the production of Aβ1-42, improving the learning and memory ability of mice and slowing down the pathological process of AD [54]. Some other studies have shown that the hyperphosphorylation of ATF6 can activate the death-associated protein kinase 1 signaling pathway, promote the phosphorylation of Bcl-2, activate autophagy, and accelerate apoptosis [47][55].
Abnormal ERS mechanisms are not only associated with AD, but also with other neurodegenerative diseases, such as prion diseases, Parkinson’s disease, and amyotrophic lateral sclerosis [47]. Consequently, the targeting of ERS in AD may be an interesting therapeutic approach, which can help us to further understand the pathogenesis of AD and provide us with a novel direction to prevent and treat AD in terms of the regulation mechanisms of ERS.
2.2. Autophagy–Lysosomal System and AD
Autophagy, also known as “self-eating”, is a highly conserved lysosomal degradation pathway that is responsible for the delivery and digestion of cellular contents, organelles and misfolded proteins in the cellular catabolic processes [60][61][62]. Based on the different degradation mechanisms [63], autophagy is classified into three general types in most mammalian cells: macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) [3][60][64]. Moreover, it should be noted that the three forms of autophagy are not exactly the same, though they share similar functions [61], which are summarized as follows: (1) macroautophagy in which the cytoplasmic component is engulfed by autophagy vacuoles and degraded by proteases after fusion with lysosomes; (2) microautophagy in which the cytoplasmic components are directly engulfed by lysosome through invagination or protrusion; and (3) CMA in which the cytoplasmic proteins are selectively delivered into lysosome by recognizing their specific motifs through lysosomal receptors [65]. Among these, macroautophagy, simply referred to as autophagy, represents the vast majority of autophagic processes [60][66]. Different from microautophagy and CMA that mainly degrade small molecules [67], macroautophagy refers to a degradation pathway that digests large protein aggregates or damaged organelles [63], and is vital to organ development and cellular function [67].
Macroautophagy begins by encasing the bulk cytoplasm or selected organelles with a double membrane of multiple proteins [65], which then becomes a double-membrane vesicle that engulfs the protein aggregates and damages the organelles through the extension of an isolation membrane, also known as the phagophore [60]. This phagophore continues to expand and engulf intracellular cargos, while sequestering inclusions in a double membranous autophagosome [63]. The autophagosomes are formed randomly in the cytoplasm and then transported along microtubules in a dynein-dependent manner towards the microtubule-organizing center [63]. Once arrived at the center, the autophagosomes may either fuse with endosomes to generate amphisomes, which may eventually merge with lysosomes to dispose of their cargo; or they may fuse directly with lysosomes to form autolysosomes [60], and then be degraded by the specific proteolytic enzymes in the lysosomes. Subsequently, the lysosomal permeases and transporters export amino acids and other by-products of degradation back to the cytoplasm for the synthesis of macromolecules, thus participating in metabolisms [63][65].
Autophagy, as a complementary mechanism for the proteasome system, is responsible for the elimination of misfolded proteins, damaged organelles and long-lived macromolecules by an essential lysosomal pathway in the cellular catabolic process [61][62][63][65], which exerts an essential cytoprotective mechanism in maintaining cellular homeostasis, energy balance and cellular defense [66][68] Although autophagy is present in all cell types, it is more important to neurons [67], as these cells are more sensitive and active to the stresses caused by damaged organelles or misfolded proteins than somatic cells, and are not easily regenerated once being eliminated [66]. Therefore, autophagy is now recognized as one of the contributors to neuronal survival and death in neurodegenerative diseases, and particularly, mounting evidence has implicated that autophagy dysregulation may play a critical role in the pathogenesis of AD [63][64]. Generally speaking, in a normal physiological state, autophagy vesicles cooperate with lysosomes to degrade abnormal proteins in healthy neurons [69]; however, in the early stage of AD, the mass production of abnormal proteins has been shown to cause damage to the autophagy–lysosome pathway [65] and with the progression of AD, autophagic dysfunction occurs continuously and autophagic vesicles accumulate steadily, which further disturbs the turnover of other molecules and aggravates the neuronal dysfunctions in AD [65]. Furthermore, autophagy dysfunctions lead to the over-accumulation of Aβ and p-Tau protein in neurons, which might directly disturb neuronal homeostasis and accelerate cell apoptosis [65]. Meanwhile, it might also affect the expression and function of other important molecules such as BACE1, apolipoprotein E (ApoE) and impair mitochondria function, which may further accelerate the progress of AD [65] (Figure 3). More interestingly, some studies have shown that ahead of the formation of the SPs and NFTs, the expressions of lysosome-related components are significantly increased, suggesting that the lysosome system is activated before the pathological alteration [7][65][70].
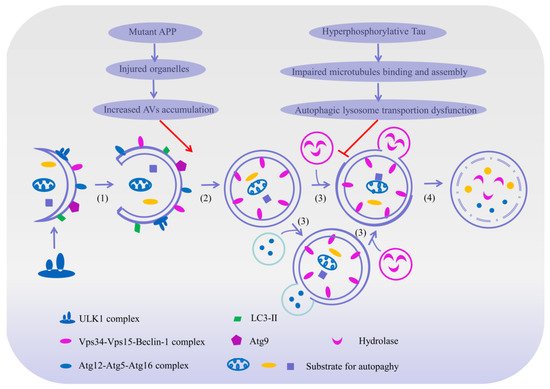
Figure 3. The mechanism of autophagy and its potential role in Alzheimer’s disease. Macroautophagy can be broken down into the following essential steps: (1) Initiation: macroautophagy begins by encasing the abnormal protein or selected organelles with an intracellular bilayer membrane structure to form a primary cup-shaped compartment containing a bilayer membrane called a phagophore. (2) Extension and completion: with the help of an Atgl2-Atg5-Atg16 complex, Atg8/LC3 and Atg9, the phagophore further engulfs the protein aggregates and impaired organelles through the extension and isolation of membranes, and finally generates a spherical double-membraned structure called an autophagosome. (3) Fusion: some autophagosomes fuse with an endosome to form an amphisome to dispose of its cargo, and others merge directly with lysosome to form an autolysosome. (4) Maturation and degradation: the amphisome and autolysosome are digested by various lysosomal hydrolases into amino acids and other small molecules, and subsequently transported back out to the cytoplasm for the synthesis of macromolecules thus taking part in metabolism. Nevertheless, the mutations of the APP gene can cause organelles’ damage, leading to an increased production of autophagy vesicles. In addition, the hyperphosphorylation of the Tau protein can impair the binding and assembly of microtubules, thereby impeding the formation and transportation of autophagosomes. When the maturation and degradation of autophagosomes are inhibited, the autophagic pathways will be damaged and a consistent accumulation of intracellular Aβ and Tau will take place, therefore possibly leading to AD. The arrow indicates the activation of process, while the T arrow indicates the inhibition of process.
The substantial evidence available manifests the idea that autophagy is involved in the processing of Aβs. It is well known that intraneuronal Aβ is generated predominantly via sequential cleavages of APP by β-secretase and γ-secretase complexes [65]. The APP belongs to the type I transmembrane protein family, which is widely distributed in various tissues, especially in the axons and dendrites of neurons [69]. The β-secretase cleaves the APP into soluble APPα and β-carboxyl-terminal fragment (β-CTF), and the γ-secretase continues to dissolve the β-CTF into various types of Aβ [71]. Yu et al. found that autophagic vacuoles (AVs) in mice hepatocytes with an overexpression of APP contained a large number of APP, β-CTF and BACE1, suggesting that AVs may be one of the potential sites for the processing of Aβs [72]. Subsequent studies have further elucidated that AVs in the brains of AD patients also contain large amounts of APP, β-CTF and γ-secretase complexes, demonstrating that autophagy was activated during the course of AD, thus leading to an amplification of AVs and production of large amounts of Aβ [72]. Consequently, all the above studies revealed that autophagy may participate in the turnover of Aβ. Thus, we have assumed that not only may the AVs degrade the encapsulated APP into Aβ, but the β-CTF in the endosome might also be delivered to the autophagosome and hydrolyzed by γ-secretase to produce more Aβ.
In addition, autophagy also takes part in the clearance of Aβ. In a physiological state, AVs that are rich in Aβs are transported retrogradely to the neuronal soma where they can fuse with the lysosome and become degraded efficiently by acidified proteases [73][74][75]; however, with the progression of AD, the hyperphosphorylation of Tau impairs microtubule binding and assembly, further impedes the AV-lysosome fusion and retrograde transportation, which in turn leads to a more rapid accumulation of AVs in the dystrophic neurites [65]. More recent studies have found that a large number of autophagosomes and other types of AVs containing APP were accumulated in the cerebral cortex and hippocampal swelling axons of AD patients, as well as model mice [76][77], indicating an impaired clearance function of AVs in AD brains. Retained AVs cannot be degraded by lysosomes effectively, which may result in Aβ accumulation in cells and accelerate the pathological processes of AD. Additionally, impairment in lysosomal membrane integrity also disrupts autophagy–lysosome function, which may further interfere with the intracellular Aβ degradation and greatly exacerbate neuron dysfunction [65][73].
References
- Cornejo, V.H.; Hetz, C. The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 2013, 35, 277–292.
- Chu, J.; Li, J.G.; Hoffman, N.E.; Madesh, M.; Praticò, D. Degradation of gamma secretase activating protein by the ubiquitin-proteasome pathway. J. Neurochem. 2015, 133, 432–439.
- Zhu, X.C.; Yu, J.T.; Jiang, T.; Tan, L. Autophagy modulation for alzheimer’s disease therapy. Mol. Neurobiol. 2013, 48, 702–714.
- Reitz, C.; Mayeux, R. Alzheimer disease: Epidemiology, diagnostic criteria, risk factors and biomarkers. Biochem. Pharmacol. 2014, 88, 640–651.
- Selkoe, D.J. Preventing alzheimer’s disease. Science 2012, 337, 1488–1492.
- Prince, Martin World Alzheimer Report. 2015. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed on 29 November 2021).
- Wu, X.L.; Piña-Crespo, J.; Zhang, Y.W.; Chen, X.C.; Xu, H.X. Tau-mediated neurodegeneration and potential implications in diagnosis and treatment of Alzheimer’s disease. Chin. Med. J. (Engl.) 2017, 130, 2978–2990.
- Shi, L.; Zhang, Z.; Li, L.; Hölscher, C. A novel dual GLP-1/GIP receptor agonist alleviates cognitive decline by re-sensitizing insulin signaling in the Alzheimer icv. STZ rat model. Behav. Brain Res. 2017, 327, 65–74.
- Budni, J.; Bellettini-Santos, T.; Mina, F.; Garcez, M.L.; Zugno, A.I. The involvement of BDNF, NGF and GDNF in aging and Alzheimer’s disease. Aging Dis. 2015, 6, 331–341.
- Ding, N.; Jiang, J.; Xu, A.; Tang, Y.; Li, Z. Manual acupuncture regulates behavior and cerebral blood flow in the SAMP8 mouse model of Alzheimer’s disease. Front. Neurosci. 2019, 13, 37.
- Plácido, A.I.; Pereira, C.M.F.; Duarte, A.I.; Candeias, E.; Correia, S.C.; Santos, R.X.; Carvalho, C.; Cardoso, S.; Oliveira, C.R.; Moreira, P.I. The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: Implications for Alzheimer’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2014, 1842, 1444–1453.
- Querfurth, H.W.; Laferla, F.M. Alzheimer’s disease: Mechanism of disease. Alzheimer’s Dis. 2010, 4, 329–344.
- Mantzavinos, V.; Alexiou, A. Biomarkers for Alzheimer’s Disease Diagnosis. Curr. Alzheimer Res. 2017, 14, 1149–1154.
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta-Mol. Basis Dis. 2011, 1812, 1584–1590.
- Kumar, D.; Gupta, S.K.; Ganeshpurkar, A.; Gutti, G.; Krishnamurthy, S.; Modi, G.; Singh, S.K. Development of Piperazinediones as dual inhibitor for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 150, 87–101.
- Li, X.; Li, N.; Sun, H.L.; Yin, J.; Tao, Y.C.; Mao, Z.X.; Yu, Z.L.; Li, W.J.; Bogden, J.D. Maternal Lead Exposure Induces Down-regulation of Hippocampal Insulin-degrading Enzyme and Nerve Growth Factor Expression in Mouse Pups. Biomed. Environ. Sci. 2017, 30, 215–219.
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease-related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305.
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 1–35.
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031.
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065.
- Gestwicki, J.E.; Garza, D. Protein Quality Control in Neurodegenerative Disease, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 107, ISBN 9780123858832.
- Chaari, A. Molecular chaperones biochemistry and role in neurodegenerative diseases. Int. J. Biol. Macromol. 2019, 131, 396–411.
- Uddin, M.S.; Tewari, D.; Sharma, G.; Kabir, M.T.; Barreto, G.E.; Bin-Jumah, M.N.; Perveen, A.; Abdel-Daim, M.M.; Ashraf, G.M. Molecular Mechanisms of ER Stress and UPR in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 2902–2919.
- Duran-Aniotz, C.; Cornejo, V.H.; Espinoza, S.; Ardiles, Á.O.; Medinas, D.B.; Salazar, C.; Foley, A.; Gajardo, I.; Thielen, P.; Iwawaki, T.; et al. IRE1 signaling exacerbates Alzheimer’s disease pathogenesis. Acta Neuropathol. 2017, 134, 489–506.
- Halliday, M.; Radford, H.; Sekine, Y.; Moreno, J.; Verity, N.; Le Quesne, J.; Ortori, C.A.; Barrett, D.A.; Fromont, C.; Fischer, P.M.; et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis. 2015, 6, e1672.
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793.
- Pereira, C.M.F. Crosstalk between Endoplasmic Reticulum Stress and Protein Misfolding in Neurodegenerative Diseases. ISRN Cell Biol. 2013, 2013, 256404.
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102.
- Ansari, N.; Khodagholi, F. Molecular Mechanism Aspect of ER Stress in Alzheimer’s Disease: Current Approaches and Future Strategies. Curr. Drug Targets 2013, 14, 114–122.
- Lee, D.Y.; Lee, K.; Lee, H.J.; Kim, D.H.; Noh, Y.H.; Yu, K.; Lee, S.H.; Lee, J.Y.; Youn, Y.C.; Jeong, Y.; et al. Activation of PERK Signaling Attenuates A b -Mediated ER Stress. PLoS ONE 2010, 5, 1–8.
- Smith, H.L.; Mallucci, G.R. The unfolded protein response: Mechanisms and therapy of neurodegeneration. Brain 2016, 139, 2113–2121.
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086.
- Futch, H.S.; Croft, C.L.; Truong, V.Q.; Krause, E.G.; Golde, T.E. Targeting psychologic stress signaling pathways in Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 49.
- Wang, J.; Chen, Y.; Zhang, C.; Xiang, Z.; Ding, J.; Han, X. Learning and memory deficits and alzheimer’s disease-like changes in mice after chronic exposure to microcystin-LR. J. Hazard. Mater. 2019, 373, 504–518.
- Ondrejcak, T.; Klyubin, I.; Hu, N.W.; Barry, A.E.; Cullen, W.K.; Rowan, M.J. Alzheimer’s disease amyloid β-protein and synaptic function. NeuroMol. Med. 2010, 12, 13–26.
- Krafft, G.A.; Klein, W.L. ADDLs and the signaling web that leads to Alzheimer’s disease. Neuropharmacology 2010, 59, 230–242.
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415.
- Cortini, F.; Roma, F.; Villa, C. Emerging roles of long non-coding RNAs in the pathogenesis of Alzheimer’s disease. Ageing Res. Rev. 2019, 50, 19–26.
- Brown, B.M.; Peiffer, J.; Rainey-Smith, S.R. Exploring the relationship between physical activity, beta-amyloid and tau: A narrative review. Ageing Res. Rev. 2019, 50, 9–18.
- Ahmed, M.R.; Shaikh, M.A.; Ul Haq, S.H.I.; Nazir, S. Neuroprotective role of chrysin in attenuating loss of dopaminergic neurons and improving motor, learning and memory functions in rats. Int. J. Health Sci. (Qassim.) 2018, 12, 35–43.
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729.
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of protein misfolding and proteostasis deficiency in protein misfolding diseases and aging. Int. J. Cell Biol. 2013, 2013, 638083.
- Jung, E.S.; Hong, H.; Kim, C.; Inhee, M.J. Acute ER stress regulates amyloid precursor protein processing through ubiquitin-dependent degradation. Sci. Rep. 2015, 5, 1–9.
- Ohno, L.D. PERK mediates eIF2α phosphorylation responsible for BACE1 elevation, CREB dysfunction and neurodegeneration in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2014, 35, 2272–2281.
- Roussel, B.D.; Kruppa, A.J.; Miranda, E.; Crowther, D.C.; Lomas, D.A.; Marciniak, S.J. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013, 12, 105–118.
- Marcora, M.S.; Belfiori-Carrasco, L.F.; Bocai, N.I.; Morelli, L.; Castaño, E.M. Amyloid-β42 clearance and neuroprotection mediated by X-box binding protein 1 signaling decline with aging in the Drosophila brain. Neurobiol. Aging 2017, 60, 57–70.
- Cai, Y.; Arikkath, J.; Yang, L.; Guo, M.L.; Periyasamy, P.; Buch, S. Interplay of endoplasmic reticulum stress and autophagy in neurodegenerative disorders. Autophagy 2016, 12, 225–244.
- Moreno, J.A.; Halliday, M.; Molloy, C.; Radford, H.; Verity, N.; Axten, J.M.; Ortori, C.A.; Willis, A.E.; Fischer, P.M.; Barrett, D.A.; et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013, 5, 206ra138.
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511.
- Erguler, K.; Pieri, M.; Deltas, C. A mathematical model of the unfolded protein stress response reveals the decision mechanism for recovery, adaptation and apoptosis. BMC Syst. Biol. 2013, 7, 16.
- Moradi Majd, R.; Mayeli, M.; Rahmani, F. Pathogenesis and promising therapeutics of Alzheimer disease through eIF2α pathway and correspondent kinases. Metab. Brain Dis. 2020, 35, 1241–1250.
- B’Chir, W.; Maurin, A.C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013, 41, 7683–7699.
- Yang, Z.; Xu, Y.; Xu, L.; Maccauro, G.; Rossi, B.; Chen, Y.; Li, H.; Zhang, J.; Sun, H.; Yang, Y.; et al. Regulation of autophagy via PERK-eIF2α effectively relieve the radiation myelitis induced by iodine-125. PLoS ONE 2013, 8, e76819.
- Du, Y.; Liu, X.; Zhu, X.; Liu, Y.; Wang, X.; Wu, X. Activating transcription factor 6 reduces Aβ1–42 and restores memory in Alzheimer’s disease model mice. Int. J. Neurosci. 2020, 130, 1015–1023.
- Sharma, R.B.; Snyder, J.T.; Alonso, L.C. Atf6α impacts cell number by influencing survival, death and proliferation. Mol. Metab. 2019, 27, S69–S80.
- Tong, Q.; Wu, L.; Jiang, T.; Ou, Z.; Zhang, Y.; Zhu, D. Inhibition of endoplasmic reticulum stress-activated IRE1α-TRAF2-caspase-12 apoptotic pathway is involved in the neuroprotective effects of telmisartan in the rotenone rat model of Parkinson’s disease. Eur. J. Pharmacol. 2016, 776, 106–115.
- Nagelkerke, A.; Bussink, J.; Sweep, F.C.G.J.; Span, P.N. The unfolded protein response as a target for cancer therapy. Biochim. Biophys. Acta-Rev. Cancer 2014, 1846, 277–284.
- Chen, L.; Xu, S.; Liu, L.; Wen, X.; Xu, Y.; Chen, J.; Teng, J. Cab45S inhibits the ER stress-induced IRE1-JNK pathway and apoptosis via GRP78/BiP. Cell Death Dis. 2014, 5, e1219.
- Palareti, G.; Legnani, C.; Cosmi, B.; Antonucci, E.; Erba, N.; Poli, D.; Testa, S.; Tosetto, A. Comparison between different D-Dimer cutoff values to assess the individual risk of recurrent venous thromboembolism: Analysis of results obtained in the DULCIS study. Int. J. Lab. Hematol. 2016, 38, 42–49.
- Cheung, Z.H.; Ip, N.Y. Autophagy deregulation in neurodegenerative diseases—Recent advances and future perspectives. J. Neurochem. 2011, 118, 317–325.
- Orr, M.E.; Oddo, S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimer’s Res. Ther. 2013, 5, 53.
- Choi, A.M.K.; Ryter, S.W.; Levine, B. Mechanisms of disease: Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662.
- Banerjee, R.; Beal, M.F.; Thomas, B. Autophagy in Neurodegenerative disorders: Pathogenic Roles and Therapeutic Implications. Trends Neurosci. 2010, 33, 541–549.
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease-locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45.
- Li, L.; Zhang, X.; Le, W. Autophagy dysfunction in Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 265–271.
- Bostancıklıoğlu, M. An update on the interactions between Alzheimer’s disease, autophagy and inflammation. Gene 2019, 705, 157–166.
- Barnett, A.; Brewer, G.J. Autophagy in aging and Alzheimer’s disease: Pathologic or protective? J. Alzheimer’s Dis. 2011, 25, 385–394.
- Liu, F.; Wang, X.Y.; Zhou, X.P.; Liu, Z.P.; Song, X.B.; Wang, Z.Y.; Wang, L. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca2+-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology 2017, 383, 13–23.
- Sodowski, K.; Cnota, W.; Czuba, B.; Borowski, D.; Wielgos, M.; Kaminski, P.; Jaczynska, R.; Wloch, A.; Kuka, D.; Zwirska-Korczala, K.; et al. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Neuroendocrinol. Lett. 2003, 115, 577–590.
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8, 84.
- Rajendran, L.; Annaert, W. Membrane Trafficking Pathways in Alzheimer’s Disease. Traffic 2012, 13, 759–770.
- Haung Yu, W.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98.
- Tammineni, P.; Ye, X.; Feng, T.; Aikal, D.; Cai, Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. eLife 2017, 6, 1–26.
- Di Domenico, F.; Tramutola, A.; Perluigi, M. Cathepsin D as a therapeutic target in Alzheimer’s disease. Expert Opin. Ther. Targets 2016, 20, 1393–1395.
- Embury, C.M.; Dyavarshetty, B.; Lu, Y.; Wiederin, J.L.; Ciborowski, P.; Gendelman, H.E.; Kiyota, T. Cathepsin B Improves ß-Amyloidosis and Learning and Memory in Models of Alzheimer’s Disease. J. Neuroimmune Pharmacol. 2017, 12, 340–352.
- Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122.
- Nixon, R.A. Autophagy, amyloidogenesis and Alzheimer disease. J. Cell Sci. 2007, 120, 4081–4091.
More
Information
Subjects:
Agriculture, Dairy & Animal Science
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
12 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No