 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Denis Jacob Machado | + 1126 word(s) | 1126 | 2020-08-21 18:27:28 | | | |
| 2 | Denis Jacob Machado | Meta information modification | 1126 | 2020-08-25 21:57:03 | | | | |
| 3 | Karina Chen | Meta information modification | 1126 | 2020-10-27 02:09:38 | | |
Video Upload Options
Responding to the ongoing and severe public health threat of viruses of the family Flaviviridae, including dengue, hepatitis C, West Nile, yellow fever, and Zika, demands a greater understanding of how these viruses emerge and spread. Updated phylogenies are central to this understanding. Most cladograms of Flaviviridae focus on specific lineages and ignore outgroups, thus hampering the efficacy of the analysis to test ingroup monophyly and relationships. This is due to the lack of annotated Flaviviridae genomes, which has gene content variation among genera. This variation makes analysis without partitioning difficult. Therefore, we developed an annotation pipeline for the genera of Flaviviridae (Flavirirus, Hepacivirus, Pegivirus, and Pestivirus), named “Fast Loci Annotation of Viruses” (FLAVi: flavi-web.com), that combines ab initio and homology-based strategies. FLAVi recovered 100% of the genes in Flavivirus and Hepacivirus genomes. In Pegivirus and Pestivirus, annotation efficiency was 100% except for one partition each. There were no false positives. The combined phylogenetic analysis of multiple genes made possible by annotation has clear impacts over the tree topology compared to phylogenies that we inferred without outgroups or data partitioning. The final tree is largely congruent with previous hypotheses and adds evidence supporting the close phylogenetic relationship between dengue and Zika.
1. Background
The family Flaviviridae comprises the genera Flavivirus, Hepacivirus, Pegivirus, and Pestivirus, all of which share structural and genomic similarity[1]. Several examples demonstrate the clinical relevance and zoonotic nature of this family. In humans, hepatitis C virus (HCV) causes a disease that damages the liver. Human pegivirus-1 (HPgV-1), causes human encephalitis[2] as well as bovine viral diarrhea (BVDV), a long-known cause of major losses in livestock[3]. Flaviviridae also houses viruses known to cause neglected tropical diseases of the genus Flavivirus, which comprises over 100 different pathogens, including notable viruses such as dengue (DENV), West Nile (WNV), yellow fever (YFV), and Zika (ZIKV)[4].
Given the medical and economic significance of Flaviviridae, there is an increasing interest in understanding their genomic structure and evolution as well as comparing known viruses of this family with less known viruses that may become a health concern in the upcoming years. However, the lack of genome annotation thwarts comparative analyses at the level of individual proteins. To overcome these shortcomings, we developed a new approach to annotate genomes from any of the four genera of Flaviviridae and evaluated its potential impact on the phylogenetic analyses of these viruses. The combined phylogenetic analysis of multiple genes made possible by annotation has a clear impacts over the tree topology compared to phylogenies that we inferred without outgroups or data partitioning. The final tree is largely congruent with previous hypotheses and adds evidence supporting the close phylogenetic relationship between dengue and Zika[5].
2. New approaches for gene prediction in Flaviviridae
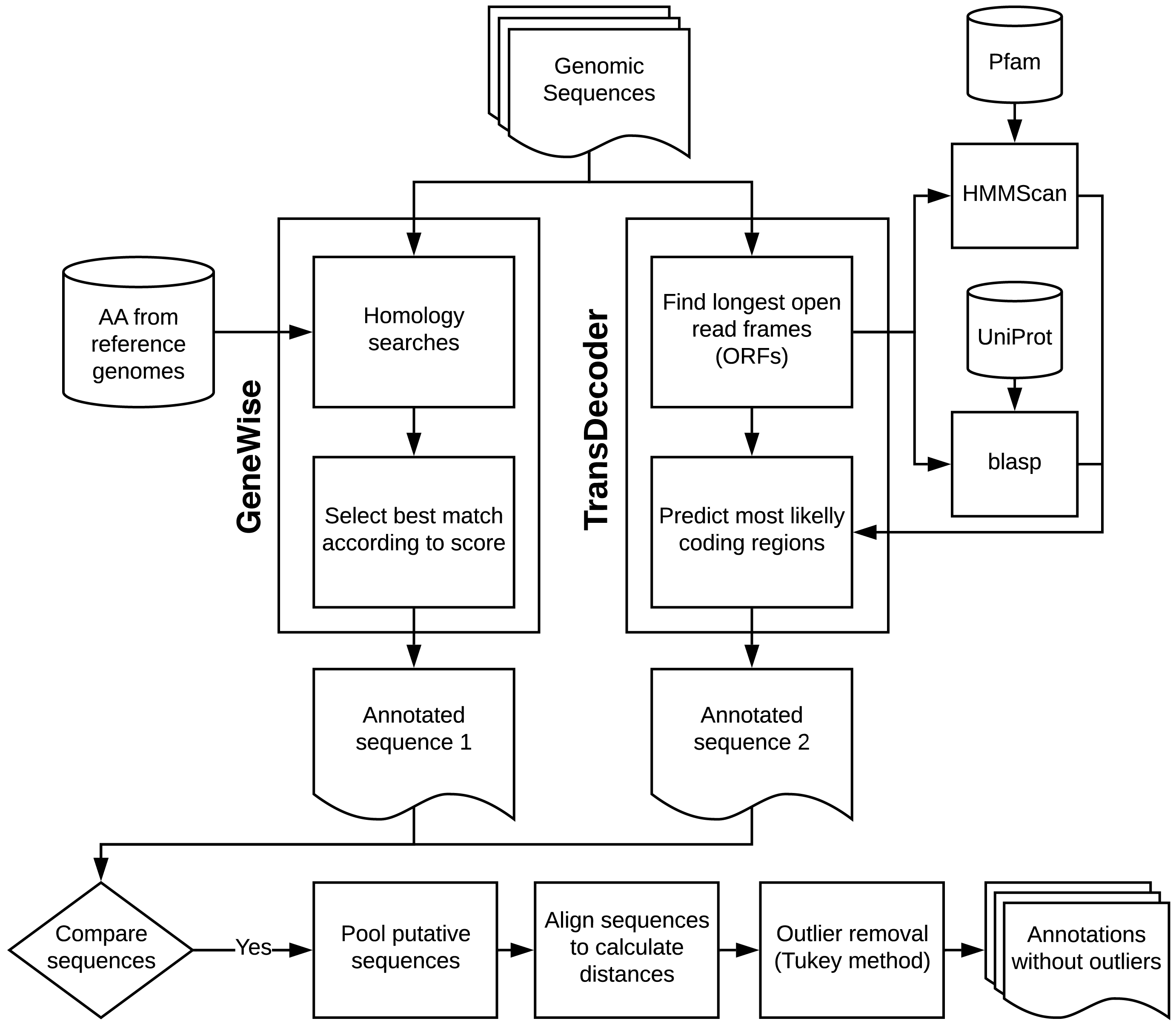
The "Fast Loci Annotation of Viruses" (FLAVi) is a new tool dedicated to annotate genomes of the family Flaviviridae and takes into account their specific characteristics (i.e. a linear and positive-sense single-stranded RNA that codes genomic polyprotein in which genes are not necessarily flanked by start and stop-codons).
Genomic sequences of Flaviviriade were processed through a pipeline that started with parallel GeneWise (Wise v2.4.1[6]) and TransDecoder v.3.0.0[7] analyses. In TransDecoder, we used dedicated databases downloaded from UniProt[8] and blastp (BLAST v2.4.0+[9]) for homology-based annotation. The user may decide to change or update the gene databases used for homology-based annotation. These databases that come with FLAVi consider all the diversity in gene content among the different genera of Flaviviridae.
The pipeline also executes ab initio analyses for different genes simultaneously, allowing the user to leverage on multiple processors. It allows the user to easily update datasets for homology searches and to train new gene models. We employed hmmscan (HMMER v3.1b2, available at hmmer.org) to search the peptides for protein domains using Pfam[10]. If predictions from both GeneWise and TransDecoder matched, we pooled the alignments and calculated distance matrices with distmat (EMBOSS v6.6.0[11]).
Finally, we applied the distance matrices for outlier testing using the Tukey method with the help of original R scripts. The Tukey method of searching for outliers leverages on the interquartile range and applies to most ranges since it is not dependent on distributional assumptions. It also ignores the mean and standard deviation, making it resistant to being influenced by the extreme values in the range. We removed all sequences that were selected as outliers.
The main steps of the gene annotation pipeline are described in the image below.
3. Web application
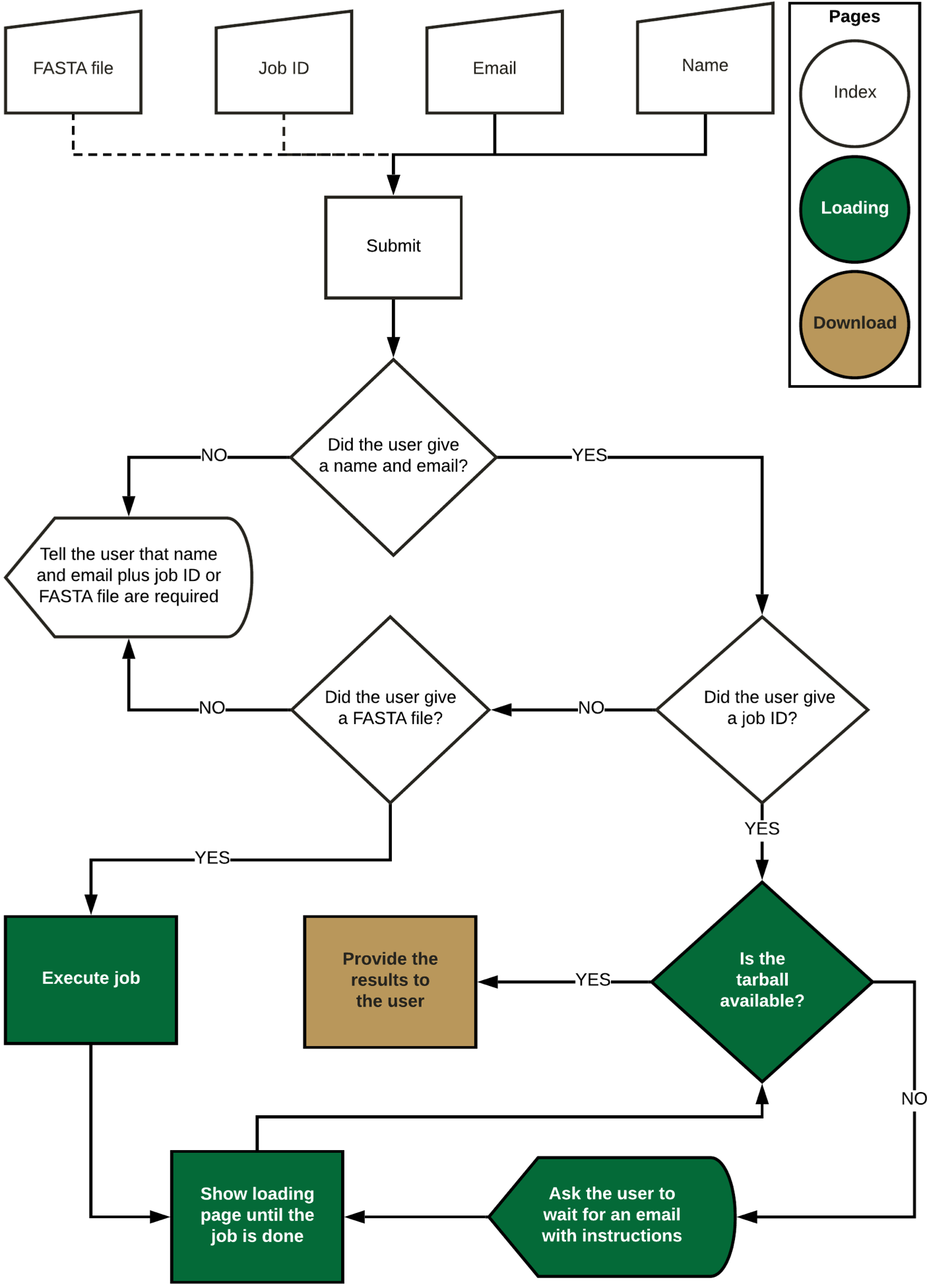
The web application is available at flavi-web.com. The main steps of the FLAVi-Web application are described in the figure below. FLAVi-Web was created using Flask, a Python-based application for the creation of web interfaces. Flask uses HTML and JavaScript to create the aesthetic properties of the web interface. FLAVi-Web was first tested on macOS Mojave with 6 CPUs and 8GB RAM. The beta version was also tested on Ubuntu with 4 CPUs and 4GB RAM. A FASTA file with the virus sequences is annotated through the FLAVi pipeline in the background to produce a tar file with the main annotation results, including the gene annotation table in GFF3 format (see software.broadinstitute.org/software/igv/GFF). The additional files, found in the compressed folder are supplementary data about each step of the annotation pipeline. These can be used for further referencing applications. During testing, a multi-FASTA file of five sequences takes about 33 min to annotate, and time increases linearly based on file size.
The accuracy and outlier tests that are part of the FLAVi pipeline are replicated within FLAVi-Web. At the time of testing, there were few annotated genomes for Flaviviridae. In NCBI's GenBank and RefSeq there were about 8,860 complete genomes (until March 2020). We have found that only 5,325 of these genomes were annotated (fully or partially). This results in ~60% of the genomes of Flaviviridae being annotated at any level. We hope that FLAVi-Web will provide annotation data to published and future genomes, adding to their usefulness in many fields of research.
4. Additional details
FLAVi was published at Viruses (DOI: 10.3390/v12080892). The new approach presented there is a dedicated annotation pipeline for Flaviviridae. Our annotation pipeline uses a combination of ab initio and homology-based strategies and recovered 100% of the genes in Flavivirus and Hepacivirus genomes. In Pegivirus and Pestivirus, annotation efficiency was 100% except for one partition each. In Pegivirus, the annotation efficiency for p7 was only 20%. In Pestivirus, FLAVi failed to recover the NS2 gene (0%). There were no false positives. So far, the pipeline was extensively tested for Flavivirus and has shown promising results for the limited number of sequences of Hepacivirus, Pegivirus, and Pestivirus that we examined, with no false-positive results. The pipeline is available at gitlab.com/MachadoDJ/FLAVi. A web application is available at flavi-web.com. Supplementary digital materials are available at Zenodo.
References
- Peter Simmonds; Paul Becher; Jens Bukh; Ernest A. Gould; Gregor Meyers; Tom Monath; Scott Muerhoff; Alexander Pletnev; Rebecca Rico-Hesse; Donald B. Smith; et al.Jack T. StapletonICTV Report Consortium ICTV Virus Taxonomy Profile: Flaviviridae. Journal of General Virology 2016, 98, 2-3, 10.1099/jgv.0.000672.
- Erin F. Balcom; Matthew A.L. Doan; William G. Branton; Juan Jovel; Gregg Blevins; Beste Edguer; Tom C Hobman; Elaine Yacyshyn; Derek Emery; Adrian Box; et al.F. K. H. Van LandeghemC. Power Human pegivirus‐1 associated leukoencephalitis: Clinical and molecular features. Annals of Neurology 2018, 84, 781-787, 10.1002/ana.25343.
- Hans Houe; Epidemiological features and economical importance of bovine virus diarrhoea virus (BVDV) infections. Veterinary Microbiology 1998, 64, 89-107, 10.1016/s0378-1135(98)00262-4.
- Michael R. Holbrook; Historical Perspectives on Flavivirus Research. Viruses 2017, 9, 97, 10.3390/v9050097.
- Machado, D. J.; Schneider, A. de B.; Guirales, S.; Janies, D.; FLAVi: An Enhanced Annotator for Viral Genomes of Flaviviridae. Viruses 2020, 12, 892, 10.3390/v12080892.
- Ewan Birney; Michele Clamp; Richard Durbin; GeneWise and Genomewise. Genome Research 2004, 14, 988-995, 10.1101/gr.1865504.
- Brian J. Haas; Alexie Papanicolaou; Moran Yassour; Manfred Grabherr; Philip D. Blood; Joshua Bowden; Matthew Brian Couger; David Eccles; Bo Li; Matthias Lieber; et al.Matthew D. MacManesMichael OttJoshua OrvisNathalie PochetFrancesco StrozziNathan T. WeeksRick WestermanThomas WilliamColin N. DeweyRobert HenschelRichard D. LeDucNir FriedmanAviv Regev De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nature Protocols 2013, 8, 1494-1512, 10.1038/nprot.2013.084.
- The UniProt Consortium; UniProt Consortium; UniProt: a worldwide hub of protein knowledge. Nucleic Acids Research 2018, 47, D506-D515, 10.1093/nar/gky1049.
- S F Altschul; Warren Gish1; Webb Miller2; Eugene W. Myers; David J. Lipman; Basic Local Alignment Search Tool. Journal of Molecular Biology 1990, 215, 403-410, 10.1006/jmbi.1990.9999.
- Sara El-Gebali; Jaina Mistry; Alex Bateman; Sean R Eddy; Aurélien Luciani; Simon C Potter; Matloob Qureshi; Lorna Richardson; Gustavo A. Salazar; Alfredo Smart; et al.Erik L L SonnhammerLayla HirshLisanna PaladinDamiano PiovesanSilvio C. E. TosattoRobert D. Finn The Pfam protein families database in 2019. Nucleic Acids Research 2018, 47, D427-D432, 10.1093/nar/gky995.
- Peter Rice; Ian Longden; Alan Bleasby; EMBOSS: The European Molecular Biology Open Software Suite. Trends in Genetics 2000, 16, 276-277, 10.1016/s0168-9525(00)02024-2.

