Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yong Weon Yi | + 2620 word(s) | 2620 | 2021-12-29 06:51:11 | | | |
| 2 | Beatrix Zheng | + 516 word(s) | 3136 | 2022-01-11 02:21:19 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yi, Y.W. Ribosomal Protein S6 as A Therapeutic Target. Encyclopedia. Available online: https://encyclopedia.pub/entry/18008 (accessed on 25 July 2026).
Yi YW. Ribosomal Protein S6 as A Therapeutic Target. Encyclopedia. Available at: https://encyclopedia.pub/entry/18008. Accessed July 25, 2026.
Yi, Yong Weon. "Ribosomal Protein S6 as A Therapeutic Target" Encyclopedia, https://encyclopedia.pub/entry/18008 (accessed July 25, 2026).
Yi, Y.W. (2022, January 11). Ribosomal Protein S6 as A Therapeutic Target. In Encyclopedia. https://encyclopedia.pub/entry/18008
Yi, Yong Weon. "Ribosomal Protein S6 as A Therapeutic Target." Encyclopedia. Web. 11 January, 2022.
Copy Citation
Ribosomal protein S6 (RPS6) is a component of the 40S small ribosomal subunit and participates in the control of mRNA translation. Additionally, phospho (p)-RPS6 has been recognized as a surrogate marker for the activated PI3K/AKT/mTORC1 pathway, which occurs in many cancer types. However, downstream mechanisms regulated by RPS6 or p-RPS remains elusive, and the therapeutic implication of RPS6 is underappreciated despite an approximately half a century history of research on this protein. In addition, substantial evidence from RPS6 knockdown experiments suggests the potential role of RPS6 in maintaining cancer cell proliferation. This motivates us to investigate the current knowledge of RPS6 functions in cancer.
ribosomal protein S6
biomarker
anticancer
cancer therapeutics
drug resistance
therapeutic target
1. RPS6 as a Therapeutic Target in Cancer
Substantial evidence suggests that RPS6 is a potential therapeutic target against cancers. Knockdown experiments have demonstrated that RPS6 itself, not only p-RPS6, is indispensable for the proliferation or survival of various cancer cells (Table 1).
Table 1. Anticancer effects of RPS6 depletion in human cancer cells.
| Cancer Type | Cell Lines | Knockdown by | Effects of RPS6-KD | Ref |
|---|---|---|---|---|
| Breast cancer, trastuzumab-resistant | JIMT-1 SK-BR-3 |
miR-129-5p mimic/siRNA |
|
[1] |
| Breast cancer, triple-negative (TNBC) | HS578T MDA-MB-231 |
siRNA |
|
[2] |
| MDA-MB-231 SUMM149PT |
siRNA |
|
[3] | |
| Cervical carcinoma | HeLa | Antisense & shRNA |
|
[4] |
| Esophageal squamous cell carcinoma (ESCC) | TE8 TE10 |
siRNA |
|
[5] |
| Gastric cancer, HER2-amplified | OE19 NCI-N87 |
siRNA |
|
[6] |
| Glioblastoma multiforme (GBM) | U251MG | siRNA |
|
[7] |
| Hepatocellular carcinoma (HCC) | SK-HEP-1 | shRNA |
|
[4] |
| Leukemia, T cell | Jurkat | antisense |
|
[4] |
| Lung cancer (adenocarcinoma) | A549 | siRNA |
|
[8] |
| Lung cancer (adenocarcinoma) | A549 | shRNA |
|
[9] |
| Lung cancer, non-small cell (NSCLC) | SK-MES-1 H1650 |
shRNA |
|
[10] |
| Lung cancer (squamous cell carcinoma) | H520 | shRNA |
|
[9] |
| Lymphoma, primary effusion lymphoma (PEL) | BC-3 | shRNA |
|
[11] |
| Lymphoma, non-Hodgkin | OCI-LY3 SUDHL-6 |
shRNA |
|
[12] |
| Melanoma, cutaneous malignant (CMM) | A375 A375VR4 SK-MEL-2 3918 |
siRNA |
|
[13] |
| Melanoma, cutaneous malignant (CMM), kinase inhibitor-resistant | A375-DR A375-TR |
siRNA |
|
[14] |
| Ovarian cancer | HEY | siRNA |
|
[15] |
| Ovarian cancer | SKOV-3 HO8910 |
siRNA |
|
[16] |
| Sarcoma, Ewing | TC32 TC71 |
siRNA |
|
[17] |
| Sarcoma, alveolar Rhabdomyosarcoma | Rh18 Rh30 |
siRNA |
|
[17] |
1.1. MicroRNAs Targeting RPS6
MiR-129-5p sensitized HER2(+) breast cancer to trastuzumab (Herceptin) by targeting RPS6 [1]. The level of miR-129-5p is reduced in the trastuzumab-resistant human breast cancer cells, JIMT-1, and patient serum samples [1][18]. MiR-129-5p reduced RPS6 expression by targeting the 3′-untranslated region (3′-UTR) of RPS6 mRNA, and transient transfection of the miR-129-5p mimic increased the trastuzumab sensitivity of JIMT-1 cells via downregulating RPS6 expression [1]. Consistent with this observation, the inhibition of miR-129-5p in the trastuzumab-susceptible breast cancer cell line, SK-BR-3, induced trastuzumab resistance, whereas concurrent treatment of RPS6-siRNA abolished this resistance. Similarly, the miR-129-5p overexpression-induced trastuzumab-sensitivity was revered by RPS6 overexpression in JIMT-1 cells [1].
1.2. RPS6-KD in Cancer Cells
RPS6 or its phosphorylation has been proposed as an alternative target to treat cancer types with high levels of RPS6 [2][19] (Figure 1). The siRNA- or shRNA-based RPS6-KD has demonstrated anticancer effects in various cancer cells (Table 1).
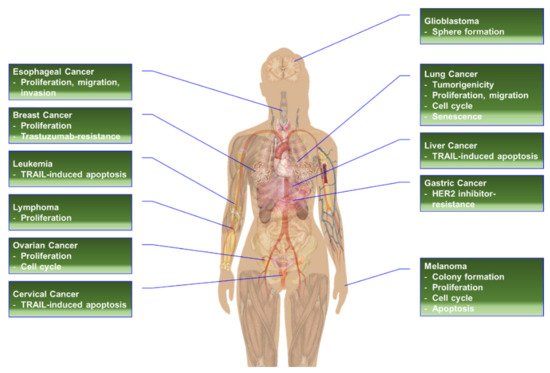
Figure 1. The effects of RPS6-KD in cancer cells.
1.2.1. RPS6-KD in Breast Cancer Cells
Studies on the TNBC cells HS578T and MDA-MB-231 have suggested RPS6 as a therapeutic target against TNBC [2]. Synergistic downregulation of both p-RPS6 (S235236) and t-RPS6 has been identified as a unique feature in TNBC cells co-treated with the EGFRi gefitinib and the METi SU11274, whereas no significant or consistent changes have been observed in the levels of p-EGFR (Y1068), t-EGFR, t-MET, p-AKT (S473), t-AKT, p-ERK1/2 (T202/Y204), and t-ERK1/2. Additionally, 4 h treatment with the proteasome inhibitor MG132 was not sufficient to inhibit the reduction of t-RPS6 in the TNBC cells in the presence of gefitinib and SU11274. The siRNA-based RPS6-KD reproduced the reduction of cell proliferation observed in TNBC cells co-treated with the gefitinib alongside SU11274. Unfortunately, the functional consequence of RPS6-KD in TNBC cells has not been further studied. However, it is noteworthy that the downregulation of t-RPS6 was augmented over time, whereas the initial reduction in the levels of p-EGFR and p-AKT was recovered with time in the TNBC cells co-treated with gefitinib and SU11274 [2]. Further research is needed to address the mechanism of regulating RPS6 stability in TNBC cells and its implication for TNBC treatment. RPS6-KD or rapamycin treatment has been reported to upregulate p-4E-BP1 (T37/46) and acetylated histone H3 (Ac-H3) in TNBC cells [3]. Co-treatment of the cells with valproic acid (VA) and tamoxifen suppresses RPS6-KD-mediated induction of p-4E-BP1 and Ac-H3 (K56) levels, and the triple combination of rapamycin, VA, and tamoxifen suppresses p-RPS6 (S235/236), p-4E-BP1 (T37/46), and Ac-H3 (K56) levels concurrently with a decrease in the viability of TNBC cells. Interestingly, this triple combination restored the expression of estrogen receptor alpha (ERα). In addition, both the triple combination and RPS6-KD with VA and tamoxifen reduced the cancer stem cell (CSC) population in TNBC cells. Furthermore, the triple combination reduced tumor growth, CSC subpopulation, and tumorigenesis in vivo [3].
1.2.2. RPS6-KD in Cervical Carcinoma Cells
Downregulation of RPS6 in the cervical carcinoma cell line HeLa resulted in the inhibition of TRAIL-dependent apoptosis in a DR4-dependent manner [4]. RPS6-KD downregulated DR4 and desensitized these cancer cells to TRAIL-dependent apoptosis (see Section 5.2.7. RPS6-KD in Hematopoietic Cancer Cells). Conversely, the overexpression of DR4 sensitized TRAIL-induced apoptosis in RPS6-KD cells. The detailed mechanism underlying the regulation of DR4 expression by RPS6 remains to be described. Similar to Jurkat cells expressing antisense RPS6, HeLa cells stably expressing antisense RPS6 or RPS6-shRNA were not resistant to apoptosis induced by doxorubicin or tunicamycin. In contrast, RPS6 overexpression in HeLa cells resulted in sensitization of the cells to TRAIL-dependent apoptosis, but not to apoptosis induced by doxorubicin, etoposide, staurosporine, or tunicamycin. Overexpression of other RPSs, such as RPS2, RPS3, and RPS20, did not affect TRAIL-induced apoptosis in HeLa cells. As mentioned earlier (see Section 3.2.3. Regulation of Apoptosis), the sensitization of TRAIL-induced apoptosis is mediated by unphosphorylated RPS6 not by p-RPS6 in HeLa cells.
1.2.3. RPS6-KD in Head and Neck Cancer Cells
The level of p-RPS6 (S240/244) and the ratio of p-RPS6/t-RPS6 are correlated with unfavorable prognosis in ESCC patients [5]. The important role of RPS6 in ESCC cells was further demonstrated by a knockdown experiment. RPS6-KD in the ESCC cell lines TE8 and TE10 reduced the numbers of these cells over time compared with the numbers of the control KD cells. Western blot analyses showed downregulation of cyclin D and CDK2 proteins in the RPS6-KD ESCC cells. In contrast, the levels of p21 and p27 were induced by RPS6-KD. Furthermore, RPS6-KD reduced the migration, invasion, and focal adhesion formation of ESCC cells, concurrently with a reduction of p-FAK (Y397), p-paxillin (Y118), p-ERK, and p-JNK (T183/Y185). S6K1-KD, an upstream kinase of RPS6, also resulted in similar effects in ESCC cells [5].
1.2.4. RPS6-KD in Gastric Cancer Cells
As mentioned earlier, RPS6-KD, in HER2i-resistant GC cell lines, resulted in a reduction in viability and induced sensitivity to HER2is through the downregulation of NRF2 and its target genes such as AKR1C1, AKR1C2, and AKR1B10 [6] (see Section 3.2.4. Regulation of Drug Resistance). The mechanisms of NRF2 downregulation by RPS6-KD remain largely unknown. Notably, RPS6 has been reported to bind to the La autoantigen (also known as Sjögren syndrome type B; SSB) upon oxidative stress, leading to increased translation of NRF2 [20]. La/SSB binds to the 5′-UTR of NRF2 mRNA in response to oxidative stress, and RPS6 consequently associates with the ribosomes.
1.2.5. RPS6-KD in Glioblastoma Cells
RPS6-KD suppressed the tumor-sphere formation of GBM cells through the inhibition of p-JAK2 and p-STAT3 and concomitantly downregulated the stemness-related proteins Nestin and SOX2 [7]. The size of the tumor spheres was also reduced by RPS6-KD. Inversely, transient overexpression of RPS6 enhanced the tumor-sphere formation of these cells [7]. Furthermore, the interleukin 6 (IL6)-induced tumor sphere formation was reduced by RPS6-KD. Taken together, RPS6 may regulate the IL6/IL6 receptor (IL6R) pathway. Since the tumor sphere formation and the expression of Nestin and SOX2 are the characteristics of GSCs, these results suggest that RPS6 plays critical roles in the development and maintenance of GSCs [7]. Interestingly, the stemness-related proteins Nestin and SOX2 have been established as transcriptional targets of STAT3 [21][22].
1.2.6. RPS6-KD in HCC Cells
Similar to HeLa cells (see Section 5.2.2. RPS6-KD in Cervical Carcinoma Cells), RPS6-KD in the human HCC cell line SK-HEP-1 suppressed TRAIL-induced apoptosis and downregulated DR4 [4]. The reconstitution of DR4 expression rescued the TRAIL-resistance in this cell line.
1.2.7. RPS6-KD in Hematopoietic Cancer Cells
A functional genetic screening for the death-rescue modifier has identified TRAIL-resistant T cell leukemia Jurkat clones that contain antisense RPS6 cDNA in their genome [4]. The reduced RPS6 expression in these cells resulted in the attenuation of TRAIL-induced apoptosis. This resistance was specific against TRAIL. No attenuation was observed in cell death caused by other signals, such as tumor necrosis factor-α (TNFα), FAS ligand (FASL), etoposide, and staurosporine.
As previously mentioned, RPS6-KD reduces the proliferation of the PEL cell line BC-3 (see Section 3.2.10. Roles of RPS6 in Response to Infection) [11]. Since BC-3 cells were infected by KSHV, the cells contain the viral genome and express the viral proteins as well [23]. RPS6 may contribute to the extraordinary stability (several days) of LANA. RPS6-KD remarkably reduced the LANA stability to 0.6 hr, leading to p53 stabilization and a p53-target gene, CDKN1A (the gene encoding p21) expression [11]. However, the molecular mechanism of RPS6 in the LANA stabilization needs further study.
RPS6-KD has decreased the levels of the proliferating cell nuclear antigen (PCNA) in p53 wild-type DLBCL cell lines [12]. However, RPS6-KD did not induce apoptosis in these cells.
1.2.8. RPS6-KD in Lung Cancer Cells
The suppression of proliferation by shRNA-based RPS6-KD was exaggerated by time in two lung cancer cell lines, A549 and H520 [9]. Inversely, the expression of senescence-associated β-galactosidase (SA-β-gal) was increased by RPS6-KD in both cell lines. Additionally, RPS6-KD increased the G0/G1 phase and decreased the G2/M phase with reduced protein levels of p-RB and cyclin D1, whereas no significant changes were observed in the levels of cyclin A, cyclin E, and total RB. More interestingly, the levels of CKIs, including p16, p21, p27, and p57, were increased. However, RPS6-KD did not induce apoptosis or altered the expression of BCL-xL, BAX, or caspase-3. The anticancer effect of RPS6-KD was reproduced in a xenograft model in which A549 lung cancer cells with RPS6-KD resulted in reduced tumorigenicity with an increase in the number of SA-β-gal(+) cells in the xenograft tissues. Similar changes were observed in the expression of cell cycle regulators and CKIs [9].
RPS6-KD induced the G0/G1 cell-cycle arrest, decreasing cell viability in the NSCLC cell lines SK-MEK-1 and H1650 [10]. The levels of p-RB, cyclin D1, cyclin E, CDK2, and CDK4 were also reduced, whereas the levels of CKIs, p21, and p27 were increased by RPS6-KD in these cells. The migration of NSCLC cells was also reduced by RPS6-KD, which also downregulated the proteins involved in cell migration, including N-cadherin, vimentin, MMP-9, MMP-2, and p-paxillin. In contrast, E-cadherin was upregulated by RPS6-KD. Inversely, the overexpression of RPS6 in a normal HBE cell line promoted cell proliferation concurrently with the upregulation of p-RPS6, p-RB, cyclin D1, cyclin E, CDK2, and CDK4, and decreases in CKIs and the number of cells in the G0/G1 phase. The migration of HBE cells was also enhanced by RPS6 overexpression. Additionally, RPS6 overexpression upregulated N-cadherin, vimentin, MMP-2, and p-paxillin in these cells [10].
As mentioned earlier (see Section 3.2.1. Regulation of Cell Cycle, Proliferation, and Growth), RPS6-KD by siRNA in A549 cells induced the p53-mediated checkpoint in an RPL11-depenent manner [8]. Although RPS6-siRNA transfection was not enough to completely deplete the RPS6 protein due to the long half-life of ribosomes, it induced the expression of p53 and p21, leading to a partial decrease in DNA synthesis and an increase in the number of cells arrested at the G1 phase. The concurrent depletion of TP53 by siRNA abrogated the effects of RPS6-siRNA transfection. RPS6 depletion increased the translation of RPL11 mRNA and the amount of ribosome-free RPL11, which can bind to MDM2, leading to the inhibition of MDM2 function to degrade p53. Consequently, the mRNAs and protein levels of p53 targets, such as CDKN1A (the gene encoding p21) and MDM2, were increased.
1.2.9. RPS6-KD in Melanoma Cells
RPS6-KD alone reduced the proliferation of two human melanoma cells A375-DR and A375-TR, which are resistant to the BRAFV600E-selective inhibitor dabrafenib and the MEK1/2 inhibitor trametinib [14]. In addition, RPS6-KD induced the G0/G1 cell-cycle arrest in these cells. More interestingly, RPS6-KD was sufficient to reduce the levels of G0/G1 checkpoint regulators such as RB, cyclin D1, and CDK6. Additionally, the enhanced downregulation of p-RPS6 by drug combinations exerted a synergistic antitumor activity [14].
In a separate study, RPS6-KD significantly reduced the colony formation and induced the apoptosis of cutaneous malignant melanoma (CMM) cells in vitro [13]. Additionally, the same authors found that S6K1 was upregulated in clinical samples with advanced stages of CMM. Furthermore, the mTORC1 signaling pathway was identified to be downregulated by the combination of the EGFRi afatinib and the ALK/ROS1 dual inhibitor crizotinib in malignant melanoma. More interestingly, the level of t-RPS6 itself, in addition to p-RPS6 level, was also downregulated by the afatinib+crizotinib treatment in melanoma cell lines in vitro [13].
1.2.10. RPS6-KD in Ovarian Cancer Cells
It has been reported that the transfection of ovarian cancer cells with RPS6-siRNA reduces the activities of glutaminase (GLS) and glutamate dehydrogenase (GDH), leading to a reduction in glutamine-induced proliferation of the cells [15]. Since glutamine is the main nutrient used by cancer cells, targeting the glutamine metabolism has been recognized as a promising therapeutic alternative [24]. Glutamine depletion inhibits cell proliferation concurrently with the induction of G1 cell-cycle arrest and apoptosis in ovarian cancer cells [15]. Glutamate increases cell proliferation in the presence of glucose. Interestingly, rapamycin reduced the glutamine-induced p-RPS6 (S235/236) and GLS expression in a dose-dependent manner in ovarian cancer cells. In the ovarian cancer cell line HEY, transfection of RPS6-siRNA was sufficient to deplete p-RSP6 (S235/236) although not t-RPS6 [15]. Under these conditions, RPS6-siRNA transfection reduced the glutamine-induced GDH activity and cell proliferation in HEY cells, whereas no significant effect of RPS6-siRNA transfection was observed in the absence of glutamine. Notably, rapamycin treatment only partially mimicked the effects of RPS6-siRNA transfection. The significance of this discrepancy remains to be elucidated.
RPS6-KD also inhibits the proliferation and long-term colony formation of the ovarian cancer cell lines SKOV-3 and HO-8910 [16]. The migration and invasion of these ovarian cancer cells are also reduced by RPS6-KD. Additionally, RPS6-KD induces the G0/G1 cell-cycle arrest. Consistently, the G0/G1 checkpoint regulators, such as cyclin D1, cyclin E, CDK2, CDK4, CDK6, and p-RB, are diminished by RPS6-KD in ovarian cancer cells [16].
1.2.11. RPS6-KD in Sarcoma Cells
An siRNA-based screening identified RPS6 as an effector of IGF1R inhibition in sarcoma cells [17]. RPS6-KD by siRNA reduced the survival of Ewing sarcoma cell lines over time. Interestingly, MTOR-KD recapitulated RPS6-KD, whereas TSC1-KD had little or no effect. In addition, RPS6-KD also induced cell death in rhabdomyosarcoma (RMS) cell lines [17]. The significance of p-RPS6 in these sarcoma cell lines is underlined by their sensitivity to the IGF1R inhibitor BMS-536924. RMS cell lines were resistant to BMS-536924 compared to Ewing sarcoma cell lines. BMS-536924 failed to downregulate p-RPS6 (S235/236) and p-4EBP1 in RMS cell line, but dose-dependently downregulated p-RPS6 (S235/236) and p-4EBP1 in sensitive Ewing sarcoma cell lines. Additional siRNA screening has found that the knockdown of macrophage stimulating 1 receptor (MST1R) sensitizes RMS cell lines to BMS-536924 with concordant reduction of p-RPS6 (S235/236). These data suggest that MST1R may be an alternative activator of RPS6 in IGF1R inhibitor-resistance sarcoma cells.
2. Conclusions
In addition to its role in protein synthesis, RPS6 may activate various pathways to induce the oxidative stress response, drug resistance, cancer stem cell stemness, and cell-cycle arrest (Figure 2). Evidence from knockdown experiments supports the therapeutic potential of RPS6 targeting in various cancer cells including breast cancer, cervical carcinoma, head and neck cancer, gastric cancer, glioblastoma, HCC, hematopoietic cancer, lung cancer, melanoma, ovarian cancer, and sarcoma (Figure 1). However, the signaling pathways downstream of RPS6 are currently largely unknown. In addition, the roles of activated RPS6 in tumorigenesis still remain elusive. The positive and negative feedback and/or feedforward mechanisms and novel regulatory pathways controlled by RPS6 may provide alternative therapeutic interventions to overcome the current limitations in cancer treatment.
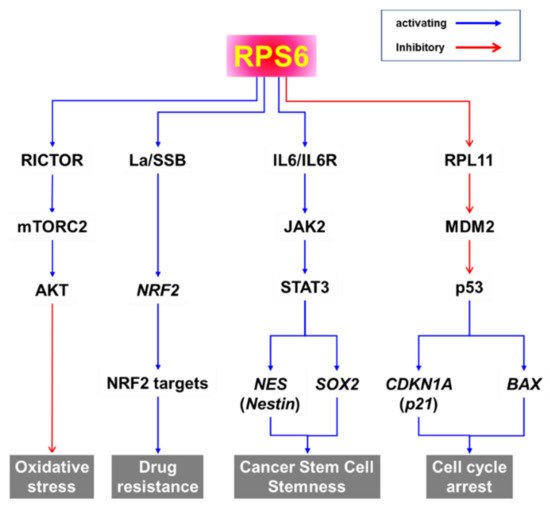
Figure 2. Putative signaling pathways downstream of RPS6.
References
- Lu, X.; Ma, J.; Chu, J.; Shao, Q.; Zhang, Y.; Lu, G.; Li, J.; Huang, X.; Li, W.; Li, Y.; et al. MiR-129-5p Sensitizes the Response of Her-2 Positive Breast Cancer to Trastuzumab by Reducing Rps6. Cell. Physiol. Biochem. 2017, 44, 2346–2356.
- Yi, Y.W.; You, K.; Bae, E.J.; Kwak, S.-J.; Seong, Y.-S.; Bae, I. Dual inhibition of EGFR and MET induces synthetic lethality in triple-negative breast cancer cells through downregulation of ribosomal protein S6. Int. J. Oncol. 2015, 47, 122–132.
- Sulaiman, A.; McGarry, S.; Lam, K.M.; El-Sahli, S.; Chambers, J.; Kaczmarek, S.; Li, L.; Addison, C.; Dimitroulakos, J.; Arnaout, A.; et al. Co-inhibition of mTORC1, HDAC and ESR1α retards the growth of triple-negative breast cancer and suppresses cancer stem cells. Cell Death Dis. 2018, 9, 815.
- Jeon, Y.-J.; Kim, I.K.; Hong, S.-H.; Nan, H.; Kim, H.-J.; Lee, H.-J.; Masuda, E.S.; Meyuhas, O.; Oh, B.-H.; Jung, Y.-K. Ribosomal protein S6 is a selective mediator of TRAIL-apoptotic signaling. Oncogene 2008, 27, 4344–4352.
- Kim, S.-H.; Jang, Y.; Chau, G.C.; Pyo, S.; Um, S.H. Prognostic significance and function of phosphorylated ribosomal protein S6 in esophageal squamous cell carcinoma. Mod. Pathol. 2012, 26, 327–335.
- Gambardella, V.; Gimeno-Valiente, F.; Tarazona, N.; Ciarpaglini, C.M.; Roda, D.; Fleitas, T.; Tolosa, P.; Cejalvo, J.M.; Huerta, M.; Roselló, S.; et al. NRF2 through RPS6 Activation Is Related to Anti-HER2 Drug Resistance in HER2-Amplified Gastric Cancer. Clin. Cancer Res. 2018, 25, 1639–1649.
- Shirakawa, Y.; Hide, T.; Yamaoka, M.; Ito, Y.; Ito, N.; Ohta, K.; Shinojima, N.; Mukasa, A.; Saito, H.; Jono, H. Ribosomal protein S6 promotes stem-like characters in glioma cells. Cancer Sci. 2020, 111, 2041–2051.
- Fumagalli, S.; Di Cara, A.; Neb-Gulati, A.; Natt, F.; Schwemberger, S.; Hall, J.; Babcock, G.F.; Bernardi, R.; Pandolfi, P.P.; Thomas, G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nature 2009, 11, 501–508.
- Chen, B.; Zhang, W.; Gao, J.; Chen, H.; Jiang, L.; Liu, D.; Cao, Y.; Zhao, S.; Qiu, Z.; Zeng, J.; et al. Downregulation of ribosomal protein S6 inhibits the growth of non-small cell lung cancer by inducing cell cycle arrest, rather than apoptosis. Cancer Lett. 2014, 354, 378–389.
- Chen, B.; Tan, Z.; Gao, J.; Wu, W.; Liu, L.; Jin, W.; Cao, Y.; Zhao, S.; Zhang, W.; Qiu, Z.; et al. Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2015, 34, 126.
- Kwun, H.J.; da Silva, S.R.; Shah, I.M.; Blake, N.; Moore, P.; Chang, Y. Kaposi’s Sarcoma-Associated Herpesvirus Latency-Associated Nuclear Antigen 1 Mimics Epstein-Barr Virus EBNA1 Immune Evasion through Central Repeat Domain Effects on Protein Processing. J. Virol. 2007, 81, 8225–8235.
- Hagner, P.R.; Mazan-Mamczarz, K.; Dai, B.; Balzer, E.M.; Corl, S.; Martin, S.S.; Zhao, X.F.; Gartenhaus, R.B. Ribosomal protein S6 is highly expressed in non-Hodgkin lymphoma and associates with mRNA containing a 5′ terminal oligopyrimidine tract. Oncogene 2010, 30, 1531–1541.
- Das, I.; Chen, H.; Maddalo, G.; Tuominen, R.; Rebecca, V.W.; Herlyn, M.; Hansson, J.; Davies, M.A.; Brage, S.E. Inhibiting insulin and mTOR signaling by afatinib and crizotinib combination fosters broad cytotoxic effects in cutaneous malignant melanoma. Cell Death Dis. 2020, 11, 882.
- Gao, M.-Z.; Wang, H.-B.; Chen, X.-L.; Cao, W.-T.; Fu, L.; Li, Y.; Quan, H.-T.; Xie, C.-Y.; Lou, L.-G. Aberrant modulation of ribosomal protein S6 phosphorylation confers acquired resistance to MAPK pathway inhibitors in BRAF-mutant melanoma. Acta Pharmacol. Sin. 2018, 40, 268–278.
- Yuan, L.; Sheng, X.; Willson, A.K.; Roque, D.R.; Stine, J.; Guo, H.; Jones, H.M.; Zhou, C.; Bae-Jump, V.L. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. -Relat. Cancer 2015, 22, 577–591.
- Yang, X.; Xu, L.; Yang, Y.-E.; Xiong, C.; Yu, J.; Wang, Y.; Lin, Y. Knockdown of ribosomal protein S6 suppresses proliferation, migration, and invasion in epithelial ovarian cancer. J. Ovarian Res. 2020, 13, 100.
- Potratz, J.C.; Saunders, D.; Wai, D.H.; Ng, T.L.; McKinney, S.E.; Carboni, J.M.; Gottardis, M.M.; Triche, T.J.; Jürgens, H.; Pollak, M.N.; et al. Synthetic Lethality Screens Reveal RPS6 and MST1R as Modifiers of Insulin-like Growth Factor-1 Receptor Inhibitor Activity in Childhood Sarcomas. Cancer Res. 2010, 70, 8770–8781.
- Luan, Q.-X.; Zhang, B.-G.; Li, X.-J.; Guo, M.-Y. MiR-129-5p is downregulated in breast cancer cells partly due to promoter H3K27m3 modification and regulates epithelial-mesenchymal transition and multi-drug resistance. Eur. Rev. Med Pharmacol. Sci. 2016, 20, 4257–4265.
- Cuesta, R.; Gupta, M.; Schneider, R.J. Chapter 7 The Regulation of Protein Synthesis in Cancer. Prog. Mol. Biol. Transl. Sci. 2009, 90, 255–292.
- Zhang, J.; Dinh, T.N.; Kappeler, K.; Tsaprailis, G.; Chen, Q.M. La Autoantigen Mediates Oxidant Induced De Novo Nrf2 Protein Translation. Mol. Cell. Proteom. 2012, 11, m111.015032.
- Zhang, C.; Mukherjee, S.; Tucker-Burden, C.; Ross, J.L.; Chau, M.J.; Kong, J.; Brat, D.J. TRIM8 regulates stemness in glioblastoma through PIAS3-STAT3. Mol. Oncol. 2017, 11, 280–294.
- Garg, M.; Shanmugam, M.K.; Bhardwaj, V.; Goel, A.; Gupta, R.; Sharma, A.; Baligar, P.; Kumar, A.P.; Goh, B.C.; Wang, L.; et al. The pleiotropic role of transcription factor STAT3 in oncogenesis and its targeting through natural products for cancer prevention and therapy. Med. Res. Rev. 2020, 41, 1291–1336.
- Arvanitakis, L.; Mesri, E.; Nador, R.; Said, J.; Asch, A.; Knowles, D.; Cesarman, E. Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus. Blood 1996, 88, 2648–2654.
- Choi, Y.-K.; Park, K.-G. Targeting Glutamine Metabolism for Cancer Treatment. Biomol. Ther. 2018, 26, 19–28.
More
Information
Subjects:
Agriculture, Dairy & Animal Science
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
11 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No