Renal hypouricemia (RHUC) is a hereditary disease that presents with increased renal urate clearance and hypouricemia due to genetic mutations in the urate transporter that reabsorbs urates in the renal proximal tubule. It is classified into the following two types: renal hypouricemia type 1 (RHUC1): A loss of function of URAT1, a transporter that uptakes urates into proximal tubule cells on the luminal side and increases renal urate clearance. As the urate clearance/creatinine clearance ratio (FEua: fractional excretion of urate) is often below 100% in RHUC1 patients, it is thought there is another unidentified urate transporter on the luminal side in addition to URAT1. Renal hypouricemia type 2 (RHUC2): The renal urate clearance increases due to a decrease in the function of GLUT9, a transporter that excretes urates at the basal side of the proximal tubule cells to the interstitium. As the urate clearance/creatinine clearance ratio of RHUC2 patients often exceeds 100% and urate is predominantly secreted, it is thought that only GLUT9 is responsible for the exit of urate reabsorption on the basal side.
1. Introduction
Exercise-induced acute kidney injury (EIAKI) is a complication of renal hypouricemia. Both renal hypouricemia types 1 and 2 have been reported to develop EIAKI
[1][2]. After several hours of exhaustive (anaerobic) exercise, loin pain, nausea and vomiting develop and serum creatinine rises a few days later as signs and symptoms of EIAKI. The change of serum creatinine varies from a mild elevation to a severe elevation requiring dialysis. Delayed computed tomography (CT) scans after the administration of contrast media have demonstrated patchy wedge-shaped enhancements suggesting renal damage due to a vascular mechanism
[3]. A renal biopsy may or may not show acute tubular necrosis. Although urate crystals are rarely found by a renal biopsy, it cannot be ruled out that renal biopsies are performed too late to detect them.
2. EIAKI Due to Increased Urate Excretion after Exhaustive Exercise
Multiple cases of EIAKI have been reported in both types 1 and 2 of renal hypouricemia; there is no significant difference in the degree of renal damage between types 1 and 2. The inhibition of an increased urinary urate excretion after exercise suppressed the onset of EIAKI in the patient with renal hypouricemia type 2 in Yeun and Hasbargen’s study and in the RHUC1 model mice. Therefore, it can be assumed that the increase in urinary urate excretion after exercise is the cause of the onset of EIAKI in both type 1 and type 2 patients with renal hypouricemia.
As RHUC type 1 is a defect in the urate uptake on the luminal side of the proximal tubule, the intracellular urate concentration in the proximal tubule cells after exercise may be lower in RHUC1 patients than in healthy subjects. As RHUC type 2 has a deficiency in the urate excretion on the basal side of the proximal tubule, the intracellular urate concentration in the proximal tubule cells after exercise may be higher in RHUC type 2 patients than in healthy subjects (Figure 1). Assuming that the mechanism of EIAKI is the same for renal hypouricemia types 1 and 2, the intracellular compartment of proximal tubule cells is unlikely to be considered to be a lesion site in terms of the intracellular urate concentration.
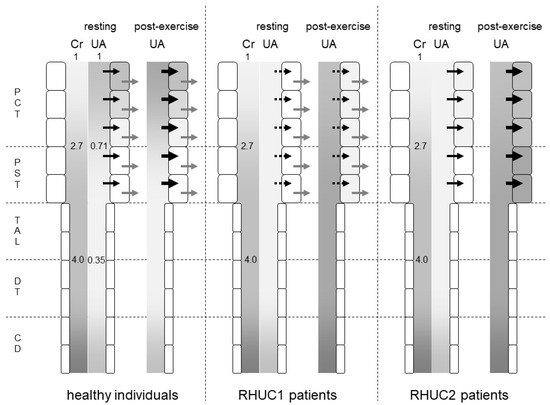
Figure 1. Predicted intraluminal/plasma concentration ratio of creatinine and urates along the nephron segments of healthy individuals, RHUC1 and RHUC2 patients at resting or post-exercise state. The gradation represents the intraluminal/plasma concentration ratio of creatinine (Cr) and urates (UAs). The darker gradation indicates a higher intraluminal/plasma concentration ratio. The intraluminal/plasma concentration ratio of Cr was predicted to be 2.7 and 4.0 at the late proximal convoluted tubule (PCT) and early distal tubule (DT), respectively. In healthy individuals, the intraluminal/plasma concentration ratio of UA was predicted to be 0.71 and 0.35 at the late PCT and early DT, respectively. Black and gray arrows represent the urate influx via URAT1 and the urate efflux via GLUT9, respectively. Large black arrows at post-exercise state indicate the enhanced urate influx via URAT1 by lactate. Dashed black arrows indicate a small urate influx via another unidentified urate transporter. At a post-exercise state, the plasma urate concentration of RHUC patients increased to reach that of healthy individuals at resting state. The darker gradation indicates that the intraluminal/plasma UA concentration ratio at the proximal straight tubule (PST) and the thick ascending limb (TAL) in RHUC patients should be higher than those in healthy individuals. The intracellular urate concentration of the proximal tubule cells in RHUC2 patients was predicted to be higher than that in RHUC1 patients. CD: collecting duct.
What is common to renal hypouricemia types 1 and 2 is impaired urate reabsorption in the proximal tubules. In the downstream nephron segments where the intraluminal urate concentration should be low in normal kidneys, the intraluminal urate concentration becomes high due to the impaired upstream urate reabsorption in the kidneys of RHUC patients. In a micropuncture study of the kidneys of Cebus monkeys
[4], the intraluminal urate concentration/plasma urate concentration ratio decreased to 0.71 ± 0.09 in the late proximal convoluted tubule and to 0.35 ± 0.05 in the early distal tubule. The intraluminal inulin concentration/plasma inulin concentration ratio increased to 2.7 ± 0.1 in the late proximal tubule and to 4.0 ± 0.2 in the early distal tubule (
Figure 1). Therefore, the FEua decreased to 0.27 ± 0.03 in the late proximal convoluted tubule and to 0.09 ± 0.01 in the early distal tubule. A total of 73% of urates in the glomerular filtrate were reabsorbed in the proximal convoluted tubule, and an additional 18% were reabsorbed in the proximal straight tubule.
Assuming that no urates are reabsorbed in the proximal tubule of RHUC2 patients, the intraluminal urate concentration at the thick ascending limb of Henle’s loop should reach about three times the blood urate concentration, as same as the intraluminal concentration of creatinine. As the serum urate level was 4.7 mg/dL when the serum creatinine level was 5.1 mg/dL in a renal hypouricemia patient
[1], the intraluminal urate concentration of the thick ascending limb reached 18.8 mg/dL. In the case of RHUC1, the reabsorption of urates other than URAT1 is thought to reduce the intraluminal urate concentration of the thick ascending limb.
In 2015, 100 µg/mL (10 mg/dL) of soluble urates were reported to stimulate Toll-like receptor 4 (TLR4) to enhance the expression of TLR4 and nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family pyrin domain-containing 3 (NLRP3) inflammasomes. The stimulated TLR4 also increased the production of caspase 1, interleukin-1β (IL-1β), and intercellular adhesion molecule 1 (ICAM-1) in cultured human proximal tubule cells
[5]. These changes were suppressed by the TLR4 inhibitor TAK242, indicating that they were mediated by TLR4 in vitro. Therefore, it was revealed that extracellular soluble urates are a ligand for TLR4.
3. TLR4 in the Kidney
TLR4 is a Toll-like receptor expressed on the cell membrane surface and a typical ligand is the lipopolysaccharide (LPS) of Gram-negative bacteria
[6]. In rat kidneys, the TLR4 molecule was reported to co-express constitutively with the Tamm–Horsfall protein in the thick ascending limb
[7]. In the kidneys of mice, the expression of TLR4 protein was reported to be on both the luminal and the basal side of the medullary thick ascending limb
[8]. However, another report demonstrated the expression of a TLR4 molecule on the luminal side of the proximal straight tubule (S3 segment) in mouse kidneys using a different anti-TLR4 antibody from the previous two studies
[9]. Therefore, TLR4 was expressed constitutively at the luminal membrane of the proximal straight tubule and/or the thick ascending limb in the kidneys.
The stimulation of the luminal TLR4 of the medullary thick ascending limb of Henle with LPS caused a signal transduction via myeloid differentiation factor 88 (MyD88)–phosphoinositide 3-kinase (PI3K)–Akt serine/threonine kinase (Akt)–mammalian target of rapamycin complex 1 (mTORC1) and suppressed the HCO
3− reabsorption of this segment
[10]. It has been reported that the stimulation of the basal TLR4 by LPS causes signal transduction via MyD88-IL-1 receptor-associated kinase 1 (IRAK1)-extracellular signal-regulated kinase (ERK) and reduced HCO
3− reabsorption
[8]. Therefore, when the intraluminal urate concentration increases after exhaustive exercise, it is considered that the MyD88-PI3K-Akt-mTORC1 pathway is activated in the cells of the proximal straight tubules and/or the thick ascending limb where the TLR4 molecule is expressed constitutively on the luminal side.
Interleukin-1β (IL-1β) is produced by the activation of the NLRP3 inflammasome
[11]. The activation of the NLRP3 inflammasome requires two steps: priming and triggering. The priming step is the expression of pro-IL-1β and NLRP3 through regulating the TLR4–inhibitor of NF-κB (IκB)–nuclear factor-κB (NF-κB) signaling and the phosphorylation of NLRP3 protein, which leads to its deubiquitination or ubiquitination of apoptosis-associated speck-like protein containing a CARD (ASC) protein to promote the assembly of NLRP3-ASC. In tubule cells, the stimulation of luminal and basal TLR4 with high concentration soluble urates after exercise may cause the expression in the priming step via NF-κB. The phosphorylation in the priming step may be caused by the basal TLR4–MyD88–IRAK1–TNF receptor-associated factor 6 (TRAF6)–TGF-β-activated kinase 1 (TAK1)–c-Jun N-terminal kinase 1 (JNK1) pathway
[12][13] (
Figure 2). As the basal TLR4–MyD88–IRAK1–ERK pathway was suppressed by the pre-treatment of the basal TLR4–Toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)–PI3K–Toll-interacting protein (Tollip)
[14], hypouricemia of RHUC patients may attenuate the suppression via the basal TLR4–TRIF–PI3K–Tollip pathway and may enhance the phosphorylation in the priming step of the NLRP3 inflammasome activation via the basal TLR4–MyD88–IRAK1–TRAF6–TAK1–JNK1 pathway.
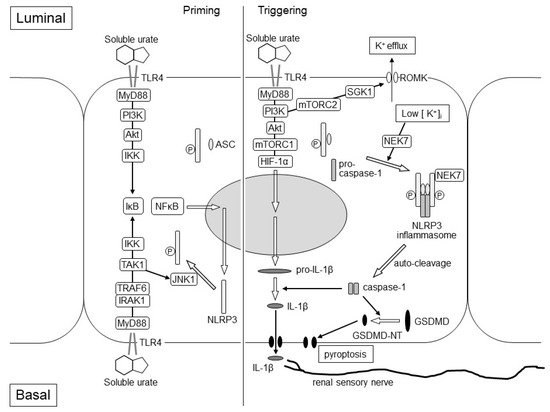
Figure 2. Hypothetical Mechanism of EIAKI in the kidneys of RHUC patients. The figure represents assumed intracellular events after TLR4 stimulation of intraluminal and basal soluble urates, which elevated transiently at the post-exhaustive exercise state of RHUC patients. The left and right parts of the figure represent the priming and triggering steps of the NLRP3 inflammasome activation, respectively. In the priming step, the expression of NLRP3 protein may be enhanced via the luminal TLR4–MyD88–PI3K–Akt–IKK–IκB–NF-κB pathway and/or the basal TLR4–MyD88–IRAK1–TRAF6–IKK–IκB–NF-κB pathway. The phosphorylation of the NLRP3 protein may be induced via the basal TLR4–MyD88–IRAK1–TRAF6–TAK1–JNK1 pathway to promote the assembly of NLRP3-ASC. In the triggering step, the NEK7-dependent assembly of the NLRP3 inflammasome with NLRP3-ASC and pro-caspase-1 may be stimulated by the luminal TLR4-MyD88-PI3K-mTORC2-SGK1 pathway, which causes K+ efflux due to the activation of the ROMK channel. Luminal TLR4 may also promote the expression of the pro-IL-1β via MyD88-PI3K-Akt-mTORC1-HIF-1α pathway. Pro-caspase-1 is auto-cleaved and converted to caspase-1, which produces matured IL-1β and GSDMD-NT from pro-IL-1β and GSDMD, respectively. IL-1β stimulates the renal sensory nerve endings around the cortical tubule, which may cause nausea, vomiting, loin pain and afferent arteriole constriction. GSDMD-NT causes membrane permeabilization and proptosis. Akt: Akt serine/threonine kinase; ASC: apoptosis-associated speck-like protein containing a CARD protein; GSDMD-NT: gasdermin D N-terminal; HIF-1α: hypoxia-inducible factor-1α; IκB: inhibitor of NF-κB; IKK: IκB kinase; IL-1β: interleukin-1β; IRAK1: IL-1 receptor-associated kinase 1; JNK1: c-Jun N-terminal kinase 1; mTORC1: mammalian target of rapamycin complex 1; mTORC2: mammalian target of rapamycin complex 2; MyD88: myeloid differentiation factor 88; NEK7: NIMA-related kinase 7; NF-κB: nuclear factor-κB; NLRP3: nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3; PI3K: phosphoinositide 3-kinase; ROMK: renal outer medulla K+ channel; SGK1: serum and glucocorticoid-inducible kinase 1; TAK1: TGF-β-activated kinase 1; TLR4: Toll-like receptor 4; TRAF6: TNF receptor-associated factor 6.
The triggering step of the NLRP3 inflammasome activation is required to form the inflammasome with NLRP3-ASC and pro-caspase-1. Stimulated luminal TLR4 also activates MyD88–PI3K–mTORC2–serum- and glucocorticoid-inducible kinase 1 (SGK1) and causes K
+ efflux due to the activation of the renal outer medulla K
+ channel (ROMK)
[15]. NIMA-related kinase 7 (NEK7) detects K
+ efflux and binds to the NLRP3 protein. NEK-bound NLRP3-ASC forms the NLRP3 inflammasome with pro-caspase-1
[16]. Therefore, K
+ efflux and the drop of intracellular K
+ concentration is a triggering signal for NLRP3 activation. (
Figure 2).
Upon activation of the NLRP3 inflammasome, pro-caspase-1 is auto-cleaved and converted to caspase-1. Caspase-1 produces inflammatory cytokines from pro-IL-1β and pro-IL-18 to matured-form IL-1β and IL-18. It also produces gasdermin D N-terminal (GSDMD-NT) from gasdermin D (GSDMD), causing membrane permeabilization and inflammation-induced cell death (pyroptosis), which would be observed pathologically as acute tubular necrosis
[17].
In the exercise loading study using RHUC1 model mice, the expression of pro-IL-1β was only enhanced in the post-exercise kidneys of the RHUC1 model mice but not in those of the control high HPRT-UoxKO mice. Through the component of the NLRP3 inflammasome, NLRP3, ASC and pro-caspase-1 were expressed both in the pre-exercise kidneys of the RHUC1 model mice and the control mice. It has also been shown that the activation of mTORC1 can promote the expression and maturation of IL-1β through hypoxia-inducible factor-1α (HIF-1α)
[18][19]. As the stimulation of the luminal TLR4 of the medullary thick ascending limb caused the signal transduction via MyD88–PI3K–Akt–mTORC1
[10], it was considered that an intraluminal high urate concentration might enhance the expression of pro-IL-1β only in the post-exercise kidneys of RHUC1 model mice (
Figure 2).
 +1 credit
+1 credit