Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | YIH-FUNG CHEN | + 3690 word(s) | 3690 | 2022-01-05 07:39:31 | | | |
| 2 | Jason Zhu | Meta information modification | 3690 | 2022-01-06 07:31:36 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chen, Y. Ion Transport System in Cervical Cancer. Encyclopedia. Available online: https://encyclopedia.pub/entry/17804 (accessed on 25 June 2026).
Chen Y. Ion Transport System in Cervical Cancer. Encyclopedia. Available at: https://encyclopedia.pub/entry/17804. Accessed June 25, 2026.
Chen, Yih-Fung. "Ion Transport System in Cervical Cancer" Encyclopedia, https://encyclopedia.pub/entry/17804 (accessed June 25, 2026).
Chen, Y. (2022, January 06). Ion Transport System in Cervical Cancer. In Encyclopedia. https://encyclopedia.pub/entry/17804
Chen, Yih-Fung. "Ion Transport System in Cervical Cancer." Encyclopedia. Web. 06 January, 2022.
Copy Citation
Cervical cancer is a significant gynecological cancer and causes cancer-related deaths worldwide. Human papillomavirus (HPV) is implicated in the etiology of cervical malignancy. However, much evidence indicates that HPV infection is a necessary but not sufficient cause in cervical carcinogenesis. Therefore, the cellular pathophysiology of cervical cancer is worthy of study.
Cl−
Ca2+
HPV
ion
1. Cell Volume Regulation and Volume-Sensitive Cl− Channels
The maintenance of homeostasis is a fundamental cellular property [1]. The regulation of cell volume, one of the fundamental cellular homeostatic mechanisms, is a widespread process that enables cells to maintain their average volume in the face of alternations in extracellular osmolarity [2]. Cells have to avoid drastic cell volume changes, which jeopardize structural integrity and constancy of the intracellular environment. Even under the constant extracellular osmolarity, cell volume is frequently challenged by the membrane transport of osmotically active substances and the formation consumption of cellular osmolytes by metabolism [3]. Accumulating evidence supports that cell volume homeostasis does not simply mean volume constancy but also serves as the integration of events in regulating cellular mechanic properties and functions, including epithelial transport, metabolism, cell proliferation, differentiation, migration, and cell death [4][5][6][7][8]. Moreover, cancer cell migration and invasion that are committed steps in tumor metastasis also involve extensive modification of the cell volume and geometrical morphology at different regions of the cells [9][10].
The homeostasis of cell volume involves the continuous functioning of the ion transport process across the plasma membrane and the fluxes of organic osmolytes and metabolites [3][11][12]. In response to the hypotonic stress, cells defend themselves by activating an efflux of cell osmolytes, including ions and specific organic molecules together with osmotically responsive water, to accomplish the process of regulatory volume decrease (RVD) [11][13]. Different ion transport systems have been reported to be responsible for the loss of K+ and Cl− in response to cell swelling [14][15]. In most cell types, the predominant pathway for RVD-associated loss of K+ and Cl− is the selective activation of separate volume-sensitive K+ and Cl− channels [2][16][17]. Several proteins have been investigated and discussed as the channel mediating the release of Cl– during RVD, e.g., ICln [18] and SWELL1 (also known as the leucine-rich repeat-containing protein 8A, LRRC8A) [19]. Another important pathway for RVD is the K+-Cl− cotransporter (KCC), which transports K+ and Cl− stoichiometrically in either direction across plasma membranes and is observed predominantly in erythrocytes [20][21][22], neurons [23], and some epithelial cells [24].
On the other hand, hypertonic stress that causes the osmotic shrinkage of cells can activate the regulatory volume increase (RVI). Shrunken cells can thereby increase their volume towards the original levels by upregulating the net influx of cell osmolytes, including Na+, Cl−, and often K+ as well, and concurrent uptake of water [25]. The central ion transport systems accomplishing electrolyte accumulation in shrunken cells are the Na+-K+-2Cl− cotransporter (NKCC) and the Na+/H+ exchanger (NHE) [26][27]. The effects of NHE also lead to the alkalization of the cell and thus the coincidental activation of the Cl−/HCO3− exchanger [28]. A schematic diagram summarizing the regulation and homeostasis of cell volume by the process of RVD and RVI is shown in Figure 1.
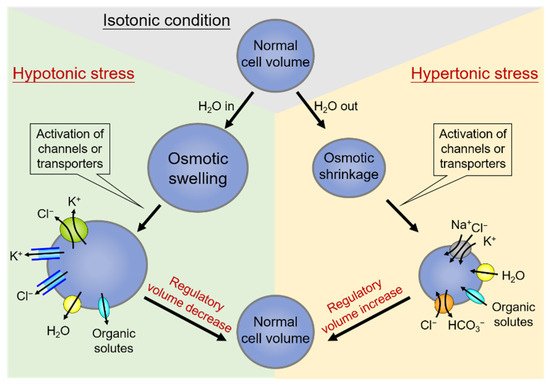
Figure 1. Regulation and homeostasis of cell volume: regulatory volume decrease (RVD) and regulatory volume increase (RVI). Under hypotonic stress conditions of cell swelling (left side), the cell activates the regulatory volume decrease (RVD). In this condition, the volume-regulatory accumulation and loss of electrolytes are mediated by the activity of membrane channels or transporters responsible for the loss of K+ and Cl−, and organic solutes along with water in response to cell swelling. On the contrary, cell volume shrinks due to water extrusion under hypertonic stress (right side), and a counter-response of regulatory volume increase (RVI) occurs to restore normal cell volume. Shrunken cells can thereby increase their volume towards the original levels by upregulating the net influx of cell osmolytes, including Na+, Cl−, and often K+, and concurrent uptake of water.
1.1. Volume-Sensitive Cl− Channels Associated with Human Cervical Carcinogenesis
Volume-regulated anion channel (VRAC) is ubiquitously expressed in vertebrate cells [29][30]. In addition to volume regulation, VRACs play essential roles in several critical physiological processes, such as osmolyte transport, metabolism, hormone release, cell migration, proliferation, and differentiation [15][31][32]. Among different VARCs, the activation of volume-sensitive Cl− channels has been demonstrated essential in the volume regulation of several non-excitable and excitable cell types [12][33]. Volume-sensitive Cl− channels have been reported to participate in cell survival and migration, given their ability to coordinate ion and water movement through the plasma membrane [2][34]. However, the expression and functional significance of volume-sensitive Cl− channels in cervical carcinoma have been less studied.
The research group was the first to study the roles of the preferentially activated Cl− channel in cervical carcinoma [35][36]. The activations of volume-sensitive Cl− currents in various human cervical epithelial cells representing different stages of cervical carcinogenesis were investigated using the whole-cell patch-clamp technique. It was found that hypotonicity activated an outward rectified, ATP-dependent, volume-sensitive Cl− current in human cervical cancer cells, including four cervical cancer cell lines, primary cells of carcinoma in situ, and invasive cancer of the cervix, but not in non-cancerous HPV-immortalized cells and normal cervical epithelial cells. This was the first report that suggested that the activation of volume-sensitive Cl− channels is associated with malignant transformation of human cervical squamous epithelium independent of various cancer stages, histopathological types, and HPV DNA positivity. Moreover, the cAMP-mediated Cl− currents were ubiquitously activated in all cervical squamous cells studied, regardless of the stages of carcinogenesis, indicating that not all of the Cl− channels are uniformly upregulated during cervical carcinogenesis [35].
In addition to the osmotic challenge, all cells possess mechanisms to maintain the homeostasis in cell volume precisely during cell cycle progression [37][38][39][40]. Especially at the G1/S transition, cells undergo a significant increase in size, disturbing the homeostasis of cell volume. Thus the process of RVD is activated to balance such cell volume decrease. It has been shown that the differential expression of K+ channels and accompanying membrane potential changes are the key to cell cycle checkpoints [41][42]. However, whether the progression of the cell cycle is accompanied by differential expression of VRAC activity was less studied. It has been previously demonstrated that cell cycle progression correlates with the expression of VRAC activity by employing the whole-cell patch-clamp recording in human cervical cancer cells under various characteristics of the cell cycle conditions [43]. The arrest of cell growth in the G0/G1 phase by aphidicolin was accompanied by a significant decrease in the VRAC current density. However, the inhibited VRAC activity was recovered by the re-entry into the cell cycle upon aphidicolin removal.
Moreover, pharmacological blockade of VRACs caused proliferating cervical cancer cells to arrest in the G0/G1 stage, indicating that activity of VRAC is critical for G1/S checkpoint progression. This was the first study that provided important information on the functional significance of VRACs in the cell cycle clock of human cervical cancer cells. These results, together with reports showing the inhibition of cell proliferation by blockage of volume-regulatory K+ and Cl− channels in other cell types, such as human peripheral T lymphocytes [44], endothelial cells [45], and microglial cells [46], indicate a possible role for these volume-sensitive channels in mitogenesis.
1.2. Differential Osmosensing Signaling Pathways of Volume-Sensitive Cl− Channels Associated with Human Cervical Carcinogenesis
The volume-sensitive Cl− channels, leading to RVD, were distinctly activated in cervical cancer cells with different tumor potentials [35][36][43][47]. One would be curious whether the osmosensing signalings involved in mediating RVD and controlling activities of volume-sensitive Cl− channels are also altered in different cervical cell types. Several signaling molecules have been suggested as potential mediators of RVD, including intracellular Ca2+, calmodulin-dependent protein kinase, protein kinase C (PKC), cyclic adenosine monophosphate (cAMP), and protein kinase A (PKA) [15][48][49][50][51]. The pharmacological screening on the possible signaling pathways involved in cell volume regulation reported that the signaling pathways mediating RVD in different cervical cell types involve the differential activation of distinct PKC isoforms [51]. Phospholipase C (MAPK) signaling with downstream activation of conventional, classic PKCs was involved in the RVD response of cervical cancer cells. On the other hand, different PKC isoforms unrelated to upstream PLC regulation were involved in the RVD of HPV-immortalized and normal cervical epithelia. Results from the whole-cell patch-clamp studies and immunofluorescence staining suggested the involvement of the conventional PKC-α, but not PKC-β or PKC-γ, in the regulation of RVD responses and activation of volume-sensitive Cl− channels in cervical cancer cells [52]. Even though the vast amount of studies demonstrated that the signaling of PKCs regulates multiple pathways relevant for cell cycle progression, tumorigenesis, and metastasis, the relevance of individual PKC isoforms in the progression of human cancer is still a matter of controversy [53]. The above-mentioned results, together with the previous studies showing the differential RVD responses in cervical cells with different malignant potentials [35][36][43], suggested the differential role of individual PKCs in cervical carcinogenesis involves the differential regulatory mechanisms on cell volume regulation and volume-sensitive Cl− channels.
The cytoskeleton is a dynamic intracellular structure that plays an essential role in regulating most biological processes, including the stability of cell shape, the onset of cell movement, and wound healing. The dynamic rearrangement of the cytoskeleton has also been shown involved in cell volume regulation in response to osmotic challenges [54][55][56]. In RVD, actin filaments are depolymerized during cell swelling, followed by actin polymerization at the phase of volume recovery. Cell swelling also increases microtubule stability and stimulates the expression of tubulin. The differential roles of actin filaments and microtubules in regulating volume-sensitive Cl− channels and RVD responses in human cervical cancer were investigated with the model of HT-3 cells [57]. The results from whole-cell voltage clamping and cell size monitoring showed that the drugs that affect cytoskeleton integrity have variable effects on the expression of volume-sensitive Cl− currents of cervical cancer cells. Depolymerization of actin with cytochalasin B potentiated the expression of Cl− currents in hypotonic stress and significantly prolonged RVD responses. In contrast, stabilization of actin polymerization by phalloidin abolished the increase in hypotonicity-elicited whole-cell Cl− conductance and retarded the cell volume recovery. Inhibition of microtubule assembly by colchicine had no effects on volume-sensitive Cl− current and RVD responses. Stabilization of microtubule by paclitaxel dose-dependently inhibited the activation of volume-sensitive Cl− channels and the process of RVD. These point to the importance of the functional integrity of actin filaments and microtubules in maintaining the effective RVD responses and activation of volume-sensitive Cl− channels in human cervical cancer cells.
However, it should be noted that the architecture of actin filaments seems to have varying effects on RVD, and volume-sensitive Cl− channels in different cell types be diverse in a cell-dependent manner, indicating the cell-type-specific requirement of actin cytoskeleton for cell-volume regulation. Some have similar findings to ours [57], showing that disruption of actin filaments potentiates the activation rate of volume-sensitive Cl− channels [58][59][60], whereas some other reports indicated that actin polymerization is required for the activation of volume-sensitive Cl− channels [61][62][63]. This also implies that different channels and transporters involved in volume regulation may have various associations with the cytoskeleton.
The above-mentioned studies on the association and osmosensing signaling of volume-sensitive Cl− channels in human cervical epithelial cells may provide a model for a better understanding of the molecular carcinogenesis of human cervical cancer. In addition, volume-sensitive Cl− channels in cervical cancer cells may offer a potential target for therapeutic intervention of cervical carcinoma and the reversal of malignant progression in human cervical carcinogenesis [64].
2. Intracellular Ca2+ Homeostasis and Store-Operated Ca2+ Entry (SOCE)
The cytosolic Ca2+ is a crucial second messenger involved in controlling diverse cellular functions, such as proliferation, differentiation, survival, migration, and gene expressions [65][66][67]. The increase in the cytosolic Ca2+ levels is mainly contributed by the Ca2+ fluxes from the extracellular space or the internal Ca2+ storage. Store-operated Ca2+ entry (SOCE), which constitutes the release of Ca2+ from the ER and the influx of Ca2+ through the plasmalemmal store-operated Ca2+ (SOC) channel, is the primary pathway to increase the cytosolic Ca2+ levels in non-excitable cells [68][69]. The molecular determinants underlying the activation of SOCE comprise two families of proteins, the ER Ca2+sensors, stromal interaction molecule 1 (STIM1) and STIM2, and the pore-forming proteins of the SOC channel, Orai1 to Orai3 [68][70][71]. STIM proteins are the ER-resident transmembrane protein with several functional domains and protein-protein interaction motifs essential for SOCE activation (see reviews in [72][73]). Once ER Ca2+ is depleted, STIM proteins aggregate into oligomers that translocate toward the plasma membrane junctions to interact with and activate Orai proteins, allowing the Ca2+ entry. STIM molecules were identified as the microtubule-interacting protein via the direction with the microtubule-plus-end-tracking proteins EB1 and EB3 [74][75]. Several studies have demonstrated the essential roles of microtubules and microtubule-plus-end-tracking mechanisms in the translocation of STIM1 toward the ER-plasma membrane junctions and the following SOCE activation [76][77][78]. With the use of the direct stochastic optical reconstruction microscopy (dSTORM), the recent study provided the ultrastructural view into the activation, aggregation, and translocation of STIM1, as well as the interaction between STIM1, microtubules, and EBs during the dynamic process of SOCE of cervical cancer cells [79]. Upon ER Ca2+ depletion, the activated STIM1 interacted with EB1 regardless of undergoing aggregation. Moreover, EB1 silencing did not impair aggregation, but the trafficking of STIM1 to the ER-plasma membrane; and EB3 compensates for the crosstalk between STIM1 and microtubule after EB1-silencing. Results from dSTORM imaging provided novel insights into STIM1 trafficking that is independent of the aggregated state and revealed the role of the microtubule network, end-binding protein EB1, and EB3 in SOCE [79].
The details of structural insights, molecular characterization, physiological functions, pathological defects of STIM and Orai proteins, as well as their dynamic protein-protein interactions that mediated the mediate the activation of SOCE, have been extensively investigated and comprehensively reviewed [80][81][82][83][84][85][86][87][88][89][90]. Increasing evidence demonstrating the essential roles of STIM and Orai proteins have made them potential prognostic biomarkers or antitumor therapeutic targets [91][92][93][94][95][96][97]. Here researchers updated the recent advances on the importance of STIM/Orai-dependent SOCE in cervical epithelial carcinogenesis and tumor malignant behaviors and the emerging development of SOCE mechanisms as the selective therapeutic target in cervical cancer.
2.1. SOCE-Dependent Ca2+ Signaling Network in Cervical Carcinogenesis
2.1.1. Proliferation and Cell Cycle Regulation
The functional significance of STIM-mediated SOCE in cervical cancer cell proliferation was extensively studied. Investigations in human cervical cancer cells showed that cell proliferation and cell cycle progression were significantly slowed down by STIM1 silencing that was attributed to the increased expression of cyclin-dependent kinase inhibitor p21 protein and decreased levels of phosphatase Cdc25C protein [98]. Results from the intracellular Ca2+ measurement in cervical cancer cells synchronized in different cell cycle status found the fluctuating SOCE activity during cell cycle progression, in which SOCE is upregulated in G1/S transition and downregulated from S to G2/M transition [99]. Mechanistic investigations showed that the blockade of SOCE activity by pharmacological inhibitors or STIM1/Orai1 silencing resulted in the decreased phosphorylation of the cyclin-dependent kinase CDK2 and increased expression of cyclin E, leading to the cell cycle arrest in G1/S transition accompanied with autophagy [99]. Therefore, these studies established the role of SOCE mediated by the STIM1 and Orai1 as the molecular determinants responsible for the Ca2+ fluxes controlling the G1/S cell cycle checkpoint of cervical cancer cells [99]. Regarding the role of STIM2 in cervical cancer cell proliferation, results from the individual or simultaneous silencing of STIM1/STIM2 suggested that both STIM1 and STIM2 contribute to the cell proliferation [76], at least partly through the regulation of SOCE during G1/S transition [99]. Furthermore, the growth of human cervical cancer xenograft in the SCID mice was attenuated by the interference with STIM1 expression or blockade of SOCE activity, demonstrating the in vivo significance of SOCE in cell proliferation [98]. These studies highlight the important roles of the STIM-mediated SOCE pathway in controlling cervical cancer cell proliferation via the regulation of the G1/S cell cycle checkpoint.
2.1.2. Tumor Angiogenesis
Tumor angiogenesis is the process of the recruitment of a new blood vessel network by which the uncontrolled growth, expansion, and dissemination of cancer cells are sustained with the supportive microenvironment enriched in oxygen and various nutrients [100]. The functional significance of STIM1-dependent SOCE in tumor angiogenesis supporting the progression of cervical cancer was revealed from the study using the model of SiHa cervical cancer cells [98]. Results from the mouse tumor xenograft model of cervical cancer showed that STIM1 silencing or SOCE blockade resulted in a reduction in tumor neovascularization and tumor growth. Measurement of the secretions of vascular endothelial growth factor (VEGF), a potent inducer of vascular endothelial cell proliferation and migration, showed that STIM1 expression regulated VEGF-A productions from cervical cancer cells. Together with other investigations dissecting the functional roles of SOCE in vascular endothelial cells [101][102][103], it is suggested that STIM1-mediated Ca2+ machinery can be an attractive therapeutic target for strategies against tumor neovascularization.
2.1.3. Cell Migration
It has been well established that SOCE dependent Ca2+ signaling network plays a vital role in the cellular migration of both non-cancerous and cancer cells through orchestrating cytoskeletal reorganization, focal adhesions, and direct sensing [104][105]. Results from STIM1 overexpression or silencing, as well as the pharmacological blockade of SOCE in cervical cancer, showed that STIM1-mediated SOCE is crucial for the migratory capability of cervical cancer cells [77][98][106]. The molecular mechanisms by which STIM1-dependent SOCE regulates cervical cancer cell migration mainly are through the Ca2+-dependent molecules controlling the focal adhesion turnover and actomyosin contractility, including calpain protease, myosin light chain kinase (MLCK), and focal adhesion proteins protein-rich tyrosine kinase (Pyk2), focal adhesion kinase (FAK), and talin. Therefore, it is proposed that STIM1-mediated Ca2+ influx regulates the contraction of myosin II-based actomyosin via the phosphorylation of the myosin II regulatory light chain by the Ca2+-dependent MLCK [106]. Moreover, the recruitment of the active focal adhesion proteins to nascent cell adhesions at the cell front, as well as the activation of the Ca2+-sensitive protease calpain at the rear end, are dependent on STIM1 expression or activity. Therefore, by altering the focal adhesion turnover and actomyosin contractility of cancer cells, STIM1-dependent SOCE promotes tumorigenesis and tumor metastasis of cervical cancers.
2.2. Diagnostic and Prognostic Values of SOCE in Cervical Carcinogenesis
Aberrated overexpression of STIM1 or Orai1 and thus upregulated SOCE activity have been observed in several types of human cancers, including cervical cancers. STIM1 and Orai1 are overexpressed in tumor tissues when compared with non-cancerous or precancerous tissues in patients with cervical cancers [76][77][98][99]. The distinct distribution of overexpressed STIM1 was identified in the invasive tumor front of the surgical specimens of human cervical cancer [107]. The studies in human cervical cancer indicated that poorer clinical outcomes, such as larger tumor size and elevated lymph node metastasis, are correlated with STIM1 upregulation in primary tumors [98], highlighting the clinical significance of STIM1 in cervical cancer progression.
Regarding STIM2, the recent study on a limited number of surgical specimens of cervical cancer showed a decreased tumoral STIM2 expression when compared with non-cancerous epithelium, whereas a higher tumoral STIM2 level when compared with invasive tumor front [76]. The simultaneous STIM1 and STIM2 immunostaining showed that, despite the overexpression of both isoforms in tumor tissues, STIM1 is the principle ER Ca2+-sensing molecule detected in the invasive tumor front [76]. These imply that STIM1 is associated with tumor growth and invasion, whereas STIM2 is mainly correlated with tumor growth. Therefore, using the STIM1/STIM2 ratio as a marker of cervical cancer aggressiveness might be promising and worth further evaluation.
2.3. Recent Development of Therapeutics Targeting SOCE in Cervical Carcinogenesis
Given the importance of SOCE tumor biology and cancer progression, it is plausible to suggest that the blockade of STIM1/Orai1-dependent Ca2+ signaling can be a practical therapeutic approach for cervical cancer. Studies on preclinical animal models have demonstrated the potentials of several small-molecule SOCE inhibitors in cancer therapies [108][109][110][111][112]. However, these SOCE inhibitors have not been approved for clinical use for cancer therapies. For example, SKF-96365 and 2-aminoethoxydiphenyl borate (2-APB), two of the potent pharmacological blockers of SOCE, prevented the tumor growth and angiogenesis in human cervical cancer-implanted SCID mice [98]. Further evidence from the overexpression or silencing of STIM1 and Orai1 supported that in vivo anti-tumor effects of SKF-96365 or 2-APB involve the blockade of STIM1/Orai1 complex [77][98].
Due to the ubiquitous expression of STIM and Orai protein, as well as their essential roles in the human immune system, including antitumor immunity, developing cancer cell-specific SOCE modulators is essential for effective antitumor therapeutics. For example, a study in the model of human cervical cancer has suggested that the different regulatory effects on the microtubule-dependent STIM1 trafficking between non-cancerous epithelial and cancerous cells could be the key to target cancer cell-specific mechanisms of SOCE activation [77]. Reversible acetylation of α-tubulin on Lys40 is important for regulating microtubule stability and function and thus modulating cell motility [113][114][115]. The histone deacetylase 6 (HDAC6) is a unique cytosol-localized HDAC member known as a prominent α-tubulin deacetylase [116][117]. It was found that the microtubule-dependent STIM1 translocation and subsequent SOCE activation of cervical cancer cells, but not in non-cancerous epithelial cells, was abrogated upon hyperacetylation of α-tubulin by pharmacological blockade or silencing of HDAC6 [77]. Thus, the microtubule-associated HDAC6 can be a cancer-specific target of malignant phenotypes mediated by STIM1-dependent SOCE, at least for cervical cancers with upregulation of HDAC6 and STIM1.
A recent investigation demonstrated the important role of the lysosomal cysteine protease cathepsin S in regulating STIM1 trafficking [118]. It highlighted the potential of the α-ketoamide-based highly selective cathepsin S inhibitor RJW-58 in the suppression of cervical cancer cell migration and invasion of cervical cancer cells [118]. Cathepsin S, a lysosomal cysteine protease, has been reported to be associated with the degradation of the extracellular matrix, thus promoting cell migration and invasion [119]. Results of immunoprecipitation assays demonstrated that cathepsin S interacted with STIM1, which was reversed by RNAi-mediated silencing and enzymatic inhibition of cathepsin S. Analyses of confocal microscopic and super-resolution imaging indicated that cathepsin S inhibition led to STIM1 puncta accumulation in the ER and interrupted the STIM1-EB1 interaction, a critical step for STIM1 trafficking towards the cell periphery. In addition, RNAi-mediated silencing and enzymatic inhibition of cathepsin S significantly decreased SOCE and reduced the activity of downstream Ca2+-dependent effectors NFAT1 and Rac1. These results provide new insight into the potential of a highly-selective cathepsin S inhibitor RJW-58 as a promising anti-cancer treatment that targets microtubule-dependent STIM1 translocation and subsequent SOCE activation [118].
References
- Davies, K.J. Adaptive homeostasis. Mol. Aspects Med. 2016, 49, 1–7.
- Okada, Y.; Okada, T.; Sato-Numata, K.; Islam, M.R.; Ando-Akatsuka, Y.; Numata, T.; Kubo, M.; Shimizu, T.; Kurbannazarova, R.S.; Marunaka, Y.; et al. Cell Volume-Activated and Volume-Correlated Anion Channels in Mammalian Cells: Their Biophysical, Molecular, and Pharmacological Properties. Pharmacol. Rev. 2019, 71, 49–88.
- Lambert, I.H.; Hoffmann, E.K.; Pedersen, S.F. Cell volume regulation: Physiology and pathophysiology. Acta Physiol. 2008, 194, 255–282.
- Guo, M.; Pegoraro, A.F.; Mao, A.; Zhou, E.H.; Arany, P.R.; Han, Y.; Burnette, D.T.; Jensen, M.H.; Kasza, K.E.; Moore, J.R.; et al. Cell volume change through water efflux impacts cell stiffness and stem cell fate. Proc. Natl. Acad. Sci. USA 2017, 114, E8618–E8627.
- Schwab, A.; Fabian, A.; Hanley, P.J.; Stock, C. Role of Ion Channels and Transporters in Cell Migration. Physiol. Rev. 2012, 92, 1865–1913.
- Häussinger, D.; Lang, F. Cell volume and hormone action. Trends Pharmacol. Sci. 1992, 13, 371–373.
- Häussinger, D. The role of cellular hydration in the regulation of cell function. Biochem. J. 1996, 313 Pt 3, 697–710.
- Pasantes-Morales, H. Channels and Volume Changes in the Life and Death of the Cell. Mol. Pharmacol. 2016, 90, 358–370.
- Morishita, K.; Watanabe, K.; Ichijo, H. Cell volume regulation in cancer cell migration driven by osmotic water flow. Cancer Sci. 2019, 110, 2337–2347.
- Wang, M.; Yang, Y.; Han, L.; Han, S.; Liu, N.; Xu, F.; Li, F. Effect of three-dimensional ECM stiffness on cancer cell migration through regulating cell volume homeostasis. Biochem. Biophys. Res. Commun. 2020, 528, 459–465.
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277.
- Jentsch, T. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307.
- Delpire, E.; Gagnon, K.B. Water Homeostasis and Cell Volume Maintenance and Regulation. Curr. Top Membr. 2018, 81, 3–52.
- Okada, Y. Ion Channels and Transporters Involved in Cell Volume Regulation and Sensor Mechanisms. Cell Biochem. Biophys. 2004, 41, 233–258.
- Hoffmann, E.K.; Pedersen, S.F. Cell volume homeostatic mechanisms: Effectors and signalling pathways. Acta Physiol. 2010, 202, 465–485.
- Furst, J.; Gschwentner, M.; Ritter, M.; Botta, G.; Jakab, M.; Mayer, M.; Garavaglia, L.; Bazzini, C.; Rodighiero, S.; Meyer, G.; et al. Molecular and functional aspects of anionic channels activated during regulatory volume decrease in mammalian cells. Pflug. Arch. 2002, 444, 1–25.
- Jahn, M.; Rauh, O.; Fauth, T.; Buerger, C. Cell Volume Regulation in the Epidermis. Cell. Physiol. Biochem. 2021, 55, 57–70.
- Costa, R.; Remigante, A.; Civello, D.A.; Bernardinelli, E.; Szabo, Z.; Morabito, R.; Marino, A.; Sarikas, A.; Patsch, W.; Paulmichl, M.; et al. O-GlcNAcylation Suppresses the Ion Current IClswell by Preventing the Binding of the Protein ICln to alpha-Integrin. Front Cell Dev. Biol. 2020, 8, 607080.
- Bertelli, S.; Remigante, A.; Zuccolini, P.; Barbieri, R.; Ferrera, L.; Picco, C.; Gavazzo, P.; Pusch, M. Mechanisms of Activation of LRRC8 Volume Regulated Anion Channels. Cell. Physiol. Biochem. 2021, 55, 41–56.
- Rust, M.B.; Alper, S.L.; Rudhard, Y.; Shmukler, B.E.; Vicente, R.; Brugnara, C.; Trudel, M.; Jentsch, T.J.; Hübner, C.A. Disruption of erythroid K-Cl cotransporters alters erythrocyte volume and partially rescues erythrocyte dehydration in SAD mice. J. Clin. Investig. 2007, 117, 1708–1717.
- Lu, D.C.-Y.; Hannemann, A.; Wadud, R.; Rees, D.C.; Brewin, J.N.; Low, P.S.; Gibson, J.S. The role of WNK in modulation of KCl cotransport activity in red cells from normal individuals and patients with sickle cell anaemia. Pflug. Arch. 2019, 471, 1539–1549.
- Ellory, J.C.; Gibson, J.S.; Stewart, G.W. Pathophysiology of Abnormal Cell Volume in Human Red Cells. Contrib. Nephrol. 1998, 123, 220–239.
- Kahle, K.T.; Khanna, A.R.; Alper, S.L.; Adragna, N.C.; Lauf, P.K.; Sun, D.; Delpire, E. K-Cl cotransporters, cell volume homeostasis, and neurological disease. Trends Mol. Med. 2015, 21, 513–523.
- Lauf, P.K.; Adragna, N.C. K-Cl Cotransport: Properties and Molecular Mechanism. Cell. Physiol. Biochem. 2000, 10, 341–354.
- Wehner, F.; Tinel, H. Role of Na+ conductance, Na(+)-H+ exchange, and Na(+)-K(+)-2Cl- symport in the regulatory volume increase of rat hepatocytes. J. Physiol. 1998, 506 Pt 1, 127–142.
- Alexander, R.T.; Grinstein, S. Na+/H+ exchangers and the regulation of volume. Acta Physiol. 2006, 187, 159–167.
- Russell, J.M. Sodium-Potassium-Chloride Cotransport. Physiol. Rev. 2000, 80, 211–276.
- Pedersen, S.F.; Counillon, L. The SLC9A-C Mammalian Na(+)/H(+) Exchanger Family: Molecules, Mechanisms, and Physiology. Physiol. Rev. 2019, 99, 2015–2113.
- Hoffmann, E.K.; Sørensen, B.H.; Sauter, D.P.R.; Lambert, I.H. Role of volume-regulated and calcium-activated anion channels in cell volume homeostasis, cancer and drug resistance. Channels 2015, 9, 380–396.
- Okada, Y.; Sato, K.; Numata, T. Pathophysiology and puzzles of the volume-sensitive outwardly rectifying anion channel. J. Physiol. 2009, 587, 2141–2149.
- Pedersen, S.F.; Okada, Y.; Nilius, B. Biophysics and Physiology of the Volume-Regulated Anion Channel (VRAC)/Volume-Sensitive Outwardly Rectifying Anion Channel (VSOR). Pflug. Arch. 2016, 468, 371–383.
- Stutzin, A.; Hoffmann, E.K. Swelling-activated ion channels: Functional regulation in cell-swelling, proliferation and apoptosis. Acta Physiol. 2006, 187, 27–42.
- Tseng, G.N. Cell swelling increases membrane conductance of canine cardiac cells: Evidence for a volume-sensitive Cl channel. Am. J. Physiol. 1992, 262, C1056–C1068.
- Okada, Y.; Shimizu, T.; Maeno, E.; Tanabe, S.; Wang, X.; Takahashi, N. Volume-sensitive Chloride Channels Involved in Apoptotic Volume Decrease and Cell Death. J. Membr. Biol. 2006, 209, 21–29.
- Chou, C.Y.; Shen, M.R.; Wu, S.N. Volume-sensitive chloride channels associated with human cervical carcinogenesis. Cancer Res. 1995, 55, 6077–6083.
- Shen, M.-R.; Wu, S.-N.; Chou, C.-Y. Volume-sensitive chloride channels in the primary culture cells of human cervical carcinoma. Biochim. Biophys. Acta 1996, 1315, 138–144.
- Tzur, A.; Kafri, R.; LeBleu, V.S.; Lahav, G.; Kirschner, M.W. Cell Growth and Size Homeostasis in Proliferating Animal Cells. Science 2009, 325, 167–171.
- Lloyd, A.C. The regulation of cell size. Cell 2013, 154, 1194–1205.
- Bryan, A.K.; Goranov, A.; Amon, A.; Manalis, S.R. Measurement of mass, density, and volume during the cell cycle of yeast. Proc. Natl. Acad. Sci. USA 2010, 107, 999–1004.
- Cikes, M. Relationship Between Growth Rate, Cell Volume, Cell Cycle Kinetics, and Antigenic Properties of Cultured Murine Lymphoma Cells23. J. Natl. Cancer Inst. 1970, 45, 978–988.
- Lang, F.; Föller, M.; Lang, K.; Lang, P.A.; Ritter, M.; Gulbins, E.; Vereninov, A.; Huber, S.M. Ion Channels in Cell Proliferation and Apoptotic Cell Death. J. Membr. Biol. 2005, 205, 147–157.
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Role of Membrane Potential in the Regulation of Cell Proliferation and Differentiation. Stem Cell Rev. Rep. 2009, 5, 231–246.
- Shen, M.R.; Droogmans, G.; Eggermont, J.; Voets, T.; Ellory, J.C.; Nilius, B. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol. 2000, 529 Pt 2, 385–394.
- Schumacher, P.A.; Sakellaropoulos, G.; Phipps, D.J.; Schlichter, L.C. Small-conductance chloride channels in human peripheral T lymphocytes. J. Membr. Biol. 1995, 145, 217–232.
- Voets, T.; Szucs, G.; Droogmans, G.; Nilius, B. Blockers of volume-activated Cl? currents inhibit endothelial cell proliferation. Pflug. Arch. 1995, 431, 132–134.
- Schlichter, L.C.; Sakellaropoulos, G.; Ballyk, B.; Pennefather, P.S.; Phipps, D.; Schlichter, L.; Sakellaropoulos, G.; Ballyk, B.; Pennefather, P.; Phipps, D. Properties of K+ and Cl–? channels and their involvement in proliferation of rat microglial cells. Glia 1996, 17, 225–236.
- Chou, C.-Y.; Shen, M.-R.; Chen, T.-M.; Huang, K.-E. Volume-activated taurine transport is differentially activated in human cervical cancer ht-3 cells but not in human papillomavirus-immortalized z183a and normal cervical epithelial cells. Clin. Exp. Pharmacol. Physiol. 1997, 24, 935–939.
- Pasantes-Morales, H.; Cardin, V.; Tuz, K. Signaling events during swelling and regulatory volume decrease. Neurochem. Res. 2000, 25, 1301–1314.
- Vázquez-Juárez, E.; Ramos-Mandujano, G.; Hernández-Benítez, R.; Pasantes-Morales, H. On the Role of G-Protein Coupled Receptors in Cell Volume Regulation. Cell. Physiol. Biochem. 2008, 21, 001–014.
- Franco, R.; Panayiotidis, M.I.; de la Paz, L.D. Autocrine signaling involved in cell volume regulation: The role of released transmitters and plasma membrane receptors. J. Cell. Physiol. 2008, 216, 14–28.
- van der Wijk, T.; Tomassen, S.F.; de Jonge, H.R.; Tilly, B.C. Signalling mechanisms involved in volume regulation of intestinal epithelial cells. Cell Physiol. Biochem. 2000, 10, 289–296.
- Chou, C.-Y.; Shen, M.-R.; Hsu, K.-S.; Huang, H.-Y.; Lin, H.-C. Involvement of PKC-α in regulatory volume decrease responses and activation of volume-sensitive chloride channels in human cervical cancer HT-3 cells. J. Physiol. 1998, 512 Pt 2, 435–448.
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2014, 33, 5225–5237.
- Mazzochi, C.; Benos, D.J.; Smith, P.R. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am. J. Physiol. Renal. Physiol. 2006, 291, F1113–F1122.
- Di Ciano-Oliveira, C.; Thirone, A.C.; Szaszi, K.; Kapus, A. Osmotic stress and the cytoskeleton: The R(h)ole of Rho GTPases. Acta Physiol. 2006, 187, 257–272.
- Pedersen, S.F.; Hoffmann, E.K.; Mills, J.W. The cytoskeleton and cell volume regulation. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 130, 385–399.
- Shen, M.R.; Chou, C.Y.; Hsu, K.F.; Hsu, K.S.; Wu, M.L. Modulation of volume-sensitive Cl -channels and cell volume by actin filaments and microtubules in human cervical cancer HT-3 cells. Acta Physiol. Scand. 1999, 167, 215–225.
- Ando-Akatsuka, Y.; Shimizu, T.; Numata, T.; Okada, Y. Involvements of the ABC protein ABCF2 and α-actinin-4 in regulation of cell volume and anion channels in human epithelial cells. J. Cell. Physiol. 2012, 227, 3498–3510.
- Levitan, I.; Almonte, C.; Mollard, P.; Garber, S.S. Modulation of a volume-regulated chloride current by F-actin. J. Membr. Biol. 1995, 147, 283–294.
- Zhang, J.; Larsen, T.H.; Lieberman, M. F-actin modulates swelling-activated chloride current in cultured chick cardiac myocytes. Am. J. Physiol. 1997, 273, C1215–C1224.
- Shimizu, T.; Fujii, T.; Ohtake, H.; Tomii, T.; Takahashi, R.; Kawashima, K.; Sakai, H. Impaired actin filaments decrease cisplatin sensitivity via dysfunction of volume-sensitive Cl—Channels in human epidermoid carcinoma cells. J. Cell. Physiol. 2020, 235, 9589–9600.
- Burow, P.; Klapperstück, M.; Markwardt, F. Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflug. Arch. 2015, 467, 1215–1226.
- Wei, H.; Mei, Y.A.; Sun, J.T.; Zhou, H.Q.; Zhang, Z.H. Regulation of swelling-activated chloride channels in embryonic chick heart cells. Cell Res. 2003, 13, 21–28.
- Xu, R.; Wang, X.; Shi, C. Volume-regulated anion channel as a novel cancer therapeutic target. Int. J. Biol. Macromol. 2020, 159, 570–576.
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802.
- Patergnani, S.; Danese, A.; Bouhamida, E.; Aguiari, G.; Previati, M.; Pinton, P.; Giorgi, C. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int. J. Mol. Sci. 2020, 21, 8323.
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058.
- Prakriya, M.; Lewis, R.S. Store-Operated Calcium Channels. Physiol. Rev. 2015, 95, 1383–1436.
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2016, 1864, 900–906.
- Bakowski, D.; Murray, F.; Parekh, A.B. Store-Operated Ca(2+) Channels: Mechanism, Function, Pharmacology, and Therapeutic Targets. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 629–654.
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43.
- Soboloff, J.; Rothberg, B.S.; Madesh, M.; Gill, D.L. STIM proteins: Dynamic calcium signal transducers. Nat. Rev. Mol. Cell Biol. 2012, 13, 549–565.
- Grabmayr, H.; Romanin, C.; Fahrner, M. STIM Proteins: An Ever-Expanding Family. Int. J. Mol. Sci. 2020, 22, 378.
- Grigoriev, I.; Gouveia, S.; Van Der Vaart, B.; Demmers, J.; Smyth, J.T.; Honnappa, S.; Splinter, D.; Steinmetz, M.; Putney, J.; Hoogenraad, C.; et al. STIM1 Is a MT-Plus-End-Tracking Protein Involved in Remodeling of the ER. Curr. Biol. 2008, 18, 177–182.
- Honnappa, S.; Gouveia, S.M.; Weisbrich, A.; Damberger, F.F.; Bhavesh, N.S.; Jawhari, H.; Grigoriev, I.; van Rijssel, F.J.; Buey, R.M.; Lawera, A.; et al. An EB1-Binding Motif Acts as a Microtubule Tip Localization Signal. Cell 2009, 138, 366–376.
- Chen, Y.F.; Chen, L.-H.; Shen, M.-R. The distinct role of STIM1 and STIM2 in the regulation of store-operated Ca2+ entry and cellular function. J. Cell. Physiol. 2019, 234, 8727–8739.
- Chen, Y.-T.; Chen, Y.-F.; Chiu, W.-T.; Liu, K.-Y.; Liu, Y.-L.; Chang, J.-Y.; Chang, H.-C.; Shen, M.-R. Microtubule-Associated Histone Deacetylase 6 Supports the Calcium Store Sensor STIM1 in Mediating Malignant Cell Behaviors. Cancer Res. 2013, 73, 4500–4509.
- Tsai, F.-C.; Seki, A.; Yang, H.W.; Hayer, A.; Carrasco, S.; Malmersjö, S.; Meyer, T. A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 2014, 16, 133–144.
- Huang, Y.-T.; Hsu, Y.-T.; Chen, Y.-F.; Shen, M.-R. Super-Resolution Microscopy Reveals That Stromal Interaction Molecule 1 Trafficking Depends on Microtubule Dynamics. Front. Physiol. 2021, 12.
- Yeung, P.S.; Yamashita, M.; Prakriya, M. Molecular basis of allosteric Orai1 channel activation by STIM1. J. Physiol. 2020, 598, 1707–1723.
- Derler, I.; Jardin, I.; Romanin, C. Molecular mechanisms of STIM/Orai communication. Am. J. Physiol. Cell Physiol. 2016, 310, C643–C662.
- Qiu, R.; Lewis, R.S. Structural features of STIM and Orai underlying store-operated calcium entry. Curr. Opin. Cell Biol. 2019, 57, 90–98.
- Gudlur, A.; Zeraik, A.E.; Hirve, N.; Hogan, P.G. STIM calcium sensing and conformational change. J. Physiol. 2020, 598, 1695–1705.
- Trebak, M.; Putney, J.W., Jr. ORAI Calcium Channels. Physiology 2017, 32, 332–342.
- Prakriya, M.; Yeung, P.S.; Yamashita, M. An open pore structure of the Orai channel, finally. Cell Calcium 2021, 94, 102366.
- Kappel, S.; Borgström, A.; Stokłosa, P.; Dörr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019, 94, 66–73.
- Berna-Erro, A.; Woodard, G.E.; Rosado, J.A. Orais and STIMs: Physiological mechanisms and disease. J. Cell. Mol. Med. 2012, 16, 407–424.
- Feske, S. CRAC channels and disease—From human CRAC channelopathies and animal models to novel drugs. Cell Calcium 2019, 80, 112–116.
- Feske, S. CRAC channelopathies. Pflug. Arch. 2010, 460, 417–435.
- Parekh, A.B. Store-operated CRAC channels: Function in health and disease. Nat. Rev. Drug Discov. 2010, 9, 399–410.
- Chen, Y.-F.; Lin, P.-C.; Yeh, Y.-M.; Chen, L.-H.; Shen, M.-R. Store-Operated Ca2+ Entry in Tumor Progression: From Molecular Mechanisms to Clinical Implications. Cancers 2019, 11, 899.
- Chen, Y.-F.; Hsu, K.-F.; Shen, M.-R. The store-operated Ca2+ entry-mediated signaling is important for cancer spread. Biochim. Biophys. Acta. 2016, 1863, 1427–1435.
- Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. Remodeling of calcium signaling in tumor progression. J. Biomed. Sci. 2013, 20, 23.
- Roberts-Thomson, S.J.; Chalmers, S.B.; Monteith, G.R. The Calcium-Signaling Toolkit in Cancer: Remodeling and Targeting. Cold Spring Harb. Perspect. Biol. 2019, 11, a035204.
- Singh, J.; Hussain, Y.; Luqman, S.; Meena, A. Targeting Ca2+ signalling through phytomolecules to combat cancer. Pharmacol. Res. 2019, 146, 104282.
- Wei, J.; Deng, Y.; Ye, J.; Luo, Y.; Weng, J.; He, Q.; Liu, F.; Li, M.; Liang, R.; Lin, Y.; et al. Store-operated Ca2+ entry as a key oncogenic Ca2+ signaling driving tumor invasion-metastasis cascade and its translational potential. Cancer Lett. 2021, 516, 64–72.
- Alharbi, A.; Zhang, Y.; Parrington, J. Deciphering the Role of Ca2+ Signalling in Cancer Metastasis: From the Bench to the Bedside. Cancers 2021, 13, 179.
- Chen, Y.-F.; Chiu, W.T.; Chen, Y.-T.; Lin, P.-Y.; Huang, H.-J.; Chou, C.-Y.; Chang, H.-C.; Tang, M.-J.; Shen, M.-R. Calcium store sensor stromal-interaction molecule 1-dependent signaling plays an important role in cervical cancer growth, migration, and angiogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15225–15230.
- Chen, Y.W.; Chen, Y.F.; Chen, Y.T.; Chiu, W.T.; Shen, M.R. The STIM1-Orai1 pathway of store-operated Ca2+ entry controls the checkpoint in cell cycle G1/S transition. Sci. Rep. 2016, 6, 22142.
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 2020, 77, 1745–1770.
- Abdullaev, I.F.; Bisaillon, J.M.; Potier, M.; Gonzalez, J.C.; Motiani, R.K.; Trebak, M. Stim1 and Orai1 Mediate CRAC Currents and Store-Operated Calcium Entry Important for Endothelial Cell Proliferation. Circ. Res. 2008, 103, 1289–1299.
- Li, J.; Cubbon, R.M.; Wilson, L.A.; Amer, M.S.; McKeown, L.; Hou, B.; Majeed, Y.; Tumova, S.; Seymour, V.A.; Taylor, H.; et al. Orai1 and CRAC Channel Dependence of VEGF-Activated Ca2+ Entry and Endothelial Tube Formation. Circ. Res. 2011, 108, 1190–1198.
- Galeano-Otero, I.; Del Toro, R.; Khatib, A.M.; Rosado, J.A.; Ordonez-Fernandez, A.; Smani, T. SARAF and Orai1 Contribute to Endothelial Cell Activation and Angiogenesis. Front. Cell Dev. Biol. 2021, 9, 639952.
- Nielsen, N.; Lindemann, O.; Schwab, A. TRP channels and STIM/ORAI proteins: Sensors and effectors of cancer and stroma cell migration. Br. J. Pharmacol. 2014, 171, 5524–5540.
- Lu, F.; Li, Y.; Lin, S.; Cheng, H.; Yang, S. Spatiotemporal regulation of store-operated calcium entry in cancer metastasis. Biochem. Soc. Trans. 2021, 49, 2581–2589.
- Chen, Y.-T.; Chen, Y.-F.; Chiu, W.-T.; Wang, Y.-K.; Chang, H.-C.; Shen, M.-R. The ER Ca2+ sensor STIM1 regulates actomyosin contractility of migratory cells. J. Cell Sci. 2013, 126, 1260–1267.
- Chen, Y.-W.; Lai, C.-S.; Chen, Y.-F.; Chiu, W.-T.; Chen, H.-C.; Shen, M.-R. STIM1-dependent Ca2+ signaling regulates podosome formation to facilitate cancer cell invasion. Sci. Rep. 2017, 7, 11523.
- Liang, X.; Zhang, N.; Pan, H.; Xie, J.; Han, W. Development of Store-Operated Calcium Entry-Targeted Compounds in Cancer. Front. Pharmacol. 2021, 12, 1349.
- Chang, Y.; Roy, S.; Pan, Z. Store-Operated Calcium Channels as Drug Target in Gastroesophageal Cancers. Front. Pharmacol. 2021, 12, 944.
- Tiffner, A.; Derler, I. Molecular Choreography and Structure of Ca(2+) Release-Activated Ca(2+) (CRAC) and KCa2+ Channels and Their Relevance in Disease with Special Focus on Cancer. Membranes 2020, 10, 425.
- Gross, S.; Mallu, P.; Joshi, H.; Schultz, B.; Go, C.; Soboloff, J. Ca2+ as a therapeutic target in cancer. Adv. Cancer Res. 2020, 148, 233–317.
- Bruce, J.I.E.; James, A.D. Targeting the Calcium Signalling Machinery in Cancer. Cancers 2020, 12, 2351.
- Malliri, A.; Caswell, P.; Ballestrem, C.; Hurlstone, A.; Garcin, C.; Straube, A. Microtubules in cell migration. Essays Biochem. 2019, 63, 509–520.
- Gadadhar, S.; Bodakuntla, S.; Natarajan, K.; Janke, C. The tubulin code at a glance. J. Cell Sci. 2017, 130, 1347–1353.
- Janke, C.; Montagnac, G. Causes and Consequences of Microtubule Acetylation. Curr. Biol. 2017, 27, R1287–R1292.
- Narita, T.; Weinert, B.T.; Choudhary, C. Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 156–174.
- Pulya, S.; Amin, S.A.; Adhikari, N.; Biswas, S.; Jha, T.; Ghosh, B. HDAC6 as privileged target in drug discovery: A perspective. Pharmacol. Res. 2020, 163, 105274.
- Lin, H.-H.; Chen, S.-J.; Shen, M.-R.; Huang, Y.-T.; Hsieh, H.-P.; Lin, S.-Y.; Lin, C.-C.; Chang, W.W.; Chang, J.-Y. Lysosomal cysteine protease cathepsin S is involved in cancer cell motility by regulating store-operated Ca2+ entry. Biochim. Et Biophys. Acta (BBA) Bioenerg. 2019, 1866, 118517.
- McDowell, S.H.; Gallaher, S.A.; Burden, R.E.; Scott, C.J. Leading the invasion: The role of Cathepsin S in the tumour microenvironment. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118781.
More
Information
Subjects:
Oncology; Obstetrics & Gynaecology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
812
Revisions:
2 times
(View History)
Update Date:
06 Jan 2022
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No