Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Flaminia Bardanzellu | + 2048 word(s) | 2048 | 2021-12-27 06:49:15 | | | |
| 2 | Jason Zhu | Meta information modification | 2048 | 2021-12-31 02:52:38 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Bardanzellu, F. Thiamine in Hypoxic-Ischemic Encephalopathy. Encyclopedia. Available online: https://encyclopedia.pub/entry/17667 (accessed on 09 August 2026).
Bardanzellu F. Thiamine in Hypoxic-Ischemic Encephalopathy. Encyclopedia. Available at: https://encyclopedia.pub/entry/17667. Accessed August 09, 2026.
Bardanzellu, Flaminia. "Thiamine in Hypoxic-Ischemic Encephalopathy" Encyclopedia, https://encyclopedia.pub/entry/17667 (accessed August 09, 2026).
Bardanzellu, F. (2021, December 30). Thiamine in Hypoxic-Ischemic Encephalopathy. In Encyclopedia. https://encyclopedia.pub/entry/17667
Bardanzellu, Flaminia. "Thiamine in Hypoxic-Ischemic Encephalopathy." Encyclopedia. Web. 30 December, 2021.
Copy Citation
Because therapeutic hypothermia and thiamine may both act on the latent period of HIE damage, a synergistic effect of these therapeutic strategies is likely. Thiamine treatment may be especially important in mild hypoxic-ischemic encephalopathy (HIE) and in areas of the world where there is limited access to expensive hypothermia equipment.
Thiamine
hypoxic-ischemic encephalopathy
neuroprotection
newborn
1. Thiamine
Humans cannot synthesize thiamine and are reliant on exogenous sources. Tissue storage of this vitamin is very limited, thus depletion of the body stores can occur rapidly after 2–3 weeks of unbalanced nutrition in healthy individuals, and after 3–5 days in patients with chronic diseases [1][2]. The half-life of thiamine after intravenous or oral administration is short, 96 min and 154 min, respectively [3].
Thiamine, and its phosphorylated intracellular derivatives, thiamine monophosphate (TMP), TPP (also named thiamine diphosphate), and triphosphate (TTP) are water-soluble organic molecules essential for normal cellular functions, growth and development in all tissues [4]. In the human body these crucial molecules can act in a myriad of biological settings and may have either a coenzyme or a non-coenzyme action [5]. In particular, at physiological pH, free thiamine has a positive charge and a definite antioxidant and anti-inflammatory activity strictly dose related. Notably, the antioxidant activity of free thiamine seems more effective for RNS [6]. Moreover, this vitamin may affect interneuronal transmission, by its role in the generation of some neurotransmitters (e.g., acetylcholine and serotonin), and neural membranes functions, because thiamine is a structural component of mitochondria and synaptosomes. Moreover, it may foster hippocampal neurogenesis [1][7][8].
TMP instead is a neutral molecule essential for transfer of thiamine across cellular membranes [4]. TPP is synthesized in the cytosol and is transported by specific carriers in mitochondria. It has a negative charge and is the most abundant form of thiamine in the body (>80%). TPP is the main biologically active form of thiamine and, in concert with magnesium, functions as an essential coenzyme in several biochemical pathways in the brain, in particular, in the metabolism of carbohydrates, lipids, and amino acids. Specifically, in carbohydrate metabolism, TPP acts both at cytoplasmic level, in glycolysis and pentose phosphate pathways for nucleotides, glutathione and lipids/myelin synthesis, and at mitochondrial level, in oxidative phosphorylation and TCA cycle for synthesis of ATP and oxidative energy production, synthesis of amino acids and glucose-derived neurotransmitters, such as glutamic acid and GABA [2]. Of note, the tripeptide glutathione, produced by TPP in the pentose phosphate pathway has powerful antioxidant activity against both cellular ROS and RNS [9]. Additionally, TPP is able to exert a direct antioxidant effect per se, as discussed below. In mitochondria, TPP may be further phosphorylated to TTP, which has two more negative charges and thus may activate high-conductance anion/chloride channels in astrocytes and neurons and play a role in regulating cholinergic and serotonergic neurotransmission. Moreover, TTP may have a role in cell energy metabolism, via its back-transformation in TPP (Figure 1) [4].
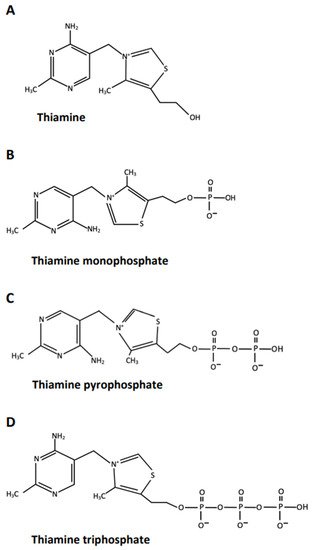
Figure 1. Chemical formula and main biological functions of thiamine and its phosphorylated derivatives. (A) Thiamine: dose related antioxidant and anti-inflammatory activity; a role in interneuronaltransmission, neural membranes function, and hippocampal neurogenesis. It is not a coenzyme; it has a positive charge. (B) Thiamine monophosphate: essential for transfer of thiamine across cellular membranes. It is not a coenzyme; it is a neutral molecule. (C) Thiamine pyrophosphate (or diphosphate): It functions as a coenzyme in several biochemical pathways in the brain, essential for synthesis of nucleotides, glutathione, lipids/myelin, ATP for oxidative energy production, several amino acids and some neurotransmitters. TPP also has a direct antioxidant activity per se. It has a negative charge. (D) Thiamine triphosphate: may activate high-conductance chloride channels; may have a role in cell energy metabolism. It is not a coenzyme, it has two more negative charges.
Thiamine requirement is related to the total caloric intake and the proportion of calories provided as carbohydrates [1]. Thus, high caloric and high carbohydrate diets increase the demand for thiamine. The recommended dose of thiamine for an average, healthy adult is 1.4 mg per day. This dose is higher in children, in critically ill conditions, in individuals with excess metabolic demand, and during pregnancy and lactation [1]. The clinical settings in which a definite TD can occur are myriad. These include a poor dietary intake, chronic alcohol misuse with malnutrition, poor gastrointestinal absorption (e.g., recurrent vomiting, chronic diarrhea, gastro-intestinal surgical procedures), increased utilization (e.g., thyrotoxicosis, chronic systemic infections, pregnancy), cancer and chemotherapeutic treatments, magnesium depletion, use of specific chemical compounds and drugs (e.g., some diuretics, tolazamide), chronic dialysis, THTR2 gene mutations [1][10][11].
Definite TD can cause serious, treatable diseases such as Wernicke’s encephalopathy (WE), peripheral neuropathy, and specific cardiovascular disturbances. In particular, severe, short-term TD commonly induces WE, whereas a mild to moderate, prolonged deficiency preferentially leads to damage to peripheral nerves [1][2]. WE is an acute life-threatening disorder fully responsive to prompt and adequate thiamine replacement. Untreated WE has an estimated mortality of 20%, and about 80% of patients who survive develop Korsakoff’s syndrome (KS), the chronic irreversible form of WE that does not remit with thiamine replacement. Of note, some cases of WE inappropriately treated with low doses of thiamine may also develop KS [1]. Importantly, the diagnosis of WE, and thiamine treatment should mandatorily be made within the first two weeks after the onset of initial symptoms/signs indicative of possible TD, during the time frame so-called “stage of reversible biochemical lesions”, when high-doses parenteral thiamine can completely reverse brain damage. Whereas, after about two weeks, irreversible structural brain lesions in specific diencephalic and brainstem areas usually occur, with consequent lack of therapeutic efficacy of thiamine replacement [1][2][12].
2. Hypoxia-Ischemia and Thiamine Deficiency in the Brain: Similar Biochemical and Histological Lesions
Past neuropathological investigations in animals and humans dating back to the 90s [13][14], and recent neuroimaging findings [15][16][17], indicate that the morphological changes in selective brain areas, such as the basal ganglia, mammillary bodies, medial thalami and the brainstem olivary complex due to TD and those due to hypoxia-ischemia may be identical. In particular, this kind of focal, selective damage in subcortical areas of the brain is a well-known distinctive pattern of WE and it is commonly observed in the majority of newborns with perinatal asphyxia when the hypoxic-ischemic insult occurs acutely. Whereas, when a subacute or chronic hypoxia-ischemia happens, which occurs in about 10% to 15% of newborns with perinatal asphyxia, this leads to preferential damage at the borders of the vascular beds of cerebral arteries. [18][19][20]. In both disorders, at the cellular level, the earliest biochemical change is likely the decrease in KGDHC enzymes activity in mitochondria of astrocytes in individuals with TD [1], and in mitochondria of astrocytes and neurons in neonates with perinatal hypoxia-ischemia [21] with consequent impairment of mitochondrial oxidative phosphorylation and, ultimately, of intracellular glucose metabolism [2]. Of note, a decrease in the activity of the other two major enzymes involved in cellular glucose metabolism, the pyruvate-dehydrogenase complex and transketolase, is noticed later after the decrease in the activity of KGDHC [1][2][21].
As a result, this acute metabolic derangement produces a global decrease in the use of glucose in the brain [1][22], with consequent severe impairment of energy metabolism in astrocytes and neurons both in HIE and in TD, with progressive activation of an excito-oxidative cascade, due to mitochondrial impairment, the occurrence of secondary subacute inflammation and eventually apoptotic cell death [1][23][24].
In both disorders, useful biomarkers of mitochondrial energy failure and a decrease in ATP production are the plasmatic increase of lactate and pyruvate levels, which in daily clinical practice indicates the opportunity to use high doses of parenteral thiamine as potential beneficial treatment.
Together, these findings are congruent with recent observations that TD per se is able to modulate the activity of the Hypoxia Inducible Factor-1α (HIF-1α) [25][26], as likewise it occurs after brain hypoxia-ischemia in full-term human neonates [27][28]. HIF-1α is the main transcription factor involved in hypoxic stress, which regulates the expression of genes involved in pro-inflammatory, pro-apoptotic and pro-survival responses [29]. The precise mechanisms underlying HIF-1α mediated neuroprotection or injury remain unclear and they may be partly related to the maturation stage of the brain at the time of the hypoxic-ischemic insult [27]. Most in vitro and in vivo findings suggest that the stabilization and activation of specific HIF-1α signaling may play an important role in promoting neuronal survival in both HIE and thiamine deficiency either by up-regulation of protective or repair genes, such as vascular-endothelial growth factor and erythropoietin, or by reduced expression of pro-apoptotic HIF-1α target genes and hence apoptotic cell death [25][26][30][27]. Of note, in TD in particular, thiamine replenishment in astrocytes is able to inhibit this specific pro-apoptotic mechanism [25][26].
3. Thiamine and Its Derivatives in the Management of Neonatal HIE
In recent years, the documented beneficial effect of TH in moderate HIE has led clinicians and researchers to evaluate and discuss cooling in combination with other pharmacologic neuroprotective agents in order to enable further improvements in outcome for infants with HIE [24][31]. However, among these compounds, thiamine has still not been considered [32][23][24][31], despite the well definite neuroprotective properties of this vitamin [2][4]. The findings extensively discussed in this review strongly suggest that thiamine given at high dosages should be included among the potential neuroprotective agents for infants with HIE, either as a complementary or an alternative treatment to TH.
This micronutrient is essential, at low concentrations, for several physiological cellular functions and development in all human tissues, in particular the brain and hearth, because of the intense metabolic demand of the nervous and cardiovascular systems [2][4]. Instead, experimental and clinical evidence indicates that very high dosages of thiamine are needed to reverse and treat the biochemical cellular lesions that may happen in pathological conditions, such as the occurrence of subacute encephalopathy/Wernicke’s encephalopathy and cardiomyopathy due to severe subacute TD [1][2].
Progressive oxidative stress in mitochondria and the cytoplasm of astrocytes and neurons, leading to excessive formation of ROS and RNS, including the very dangerous compound peroxynitrite, is likely the main pathophysiologic determinant of brain damage in HIE [33][34][24]. Researchers suggest that thiamine per se as antioxidant, given intravenous at high doses, should counteract potently the damaging effects of ROS and RNS, including the reaction of peroxynitrite with the tyrosine residues of the major enzymes involved in cellular glucose metabolism, which is the likely earliest biochemical change in HIE [1][21][4][5]. This is supported by several experimental studies on diverse oxidants which document that thiamine at high dosages is able to diminish or counteract the production of both ROS and RNS, although it is much more effective toward RNS [6][35]. Of note, among thiamine derivatives, TPP is also able to exert both a direct antioxidant effect per se and an indirect antioxidant activity, acting as an essential coenzyme, in the pentose phosphate pathway, for the synthesis of a major cellular antioxidant, the tripeptide glutathione [2]. Thus, in newborns with HIE, to boost in the brain the antioxidant defense mechanisms by increasing the levels of thiamine and TPP should limit the amount of ROS/RNS to a level that is not threatening for the integrity of lipids, proteins, carbohydrates and nucleic acids [36]. Additionally, among the myriad of possible molecular interactions between thiamine and biological compounds, of particular relevance for treatment of HIE is the well documented interaction between thiamine and peroxynitrite [6]. Indeed, thiamine may compete with the tyrosine residues of the major enzymes involved in cellular glucose metabolism to bind peroxynitrite anions (ONOO–) and activate different oxido-reductive biochemical pathways less dangerous for cell viability [6][37]. In particular, it is hypothesizable a greater affinity, at physiological pH. of the negatively charged peroxynitrite (formal charge, 1–) for the positively charged free thiamine (formal charge, 1+) compared with tyrosine which is a neutral molecule (formal charge, 0). (Figure 2) Studies on TD provide indirect support for an interaction of peroxynitrite with thiamine [6]. In particular, the finding that in neurons within susceptible brain areas deficiency of thiamine induces tyrosine nitration [38].

In addition to the well-defined role in counteracting oxidative stress, high doses of thiamine and its derivatives may have other multiple potential benefits in infants with HIE, which include fostering the activity of the major enzymes involved in intracellular glucose metabolism, a definite anti-inflammatory activity by differential regulation of pro-inflammatory mediators, a modulatory effect on interneuronal transmission, the enhancement of hippocampal neurogenesis and the maintenance of mitochondrial function by recovery of mitochondrial network architecture, membrane potential and respiration [4][5][7][39][40], thus avoiding potential mitochondrial failure, and in turn preventing neuronal and glial cell death [41].
References
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455.
- Sechi, G.; Sechi, E.; Fois, C.; Kumar, N. Advances in clinical determinants and neurological manifestations of B vitamin deficiency in adults. Nutr. Rev. 2016, 74, 281–300.
- Sechi, G. Concerning “Genetic defects of thiamine transport and metabolism: A review of clinical phenotypes, genetics and functional studies” by Marcé-Grau et al. J. Inherit. Metab. Dis. 2020, 43, 159–160.
- Manzetti, S.; Zhang, J.; van der Spoel, D. Thiamin function, metabolism, uptake, and transport. Biochemistry 2014, 53, 821–835.
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.; Kaehne, T.; Di Salvo, L.M.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorff, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 5, 12583.
- Huang, H.-M.; Chen, H.-L.; Gibson, G.E. Thiamine and oxidants interact to modify cellular calcium stores. Neurochem. Res. 2010, 35, 2107–2116.
- Ba, A. Metabolic and structural role of thiamine in nervous tissues. Cell. Mol. Neurobiol. 2008, 28, 923–931.
- Zhao, N.; Zhong, C.; Wang, Y.; Zhao, Y.; Gong, N.; Zhou, G.; Xu, T.; Hong, Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiol. Dis. 2008, 29, 176–185.
- Ramalingam, M.; Kim, S.-J. Reactive oxygen/nitrogen species and their functional correlations in neurodegenerative diseases. J. Neural Transm. 2012, 119, 891–910.
- Sechi, G.; Batzu, L.; Agrò, L.; Fois, C. Cancer-related Wernicke-Korsakoff syndrome. Lancet Oncol. 2016, 17, e221–e222.
- Sechi, E.; Addis, A.; Fadda, G.; Minafra, L.; Bravatà, V.; Sechi, G. Teaching neuroimages: Subacute encephalopathy in a young woman with THTR2 gene mutation. Neurology 2015, 85, e108–e109.
- Sechi, G.; Sechi, M.M. Challenges in prediction and diagnosis of alcohol withdrawal syndrome and Wernicke encephalopathy. JAMA Intern. Med. 2020, 180, 1716.
- Vortmeyer, A.O.; Colmant, H.J. Differentiation between brain lesions in experimental thiamine deficiency. Virchows Arch. A Pathol. Anat. Histopathol. 1988, 414, 61–67.
- Vortmeyer, A.O.; Hagel, C.; Laas, R. Hypoxia-ischemia and thiamine deficiency. Clin. Neuropathol. 1993, 12, 184–190.
- Schmidtke, K. Wernicke-Korsakoff syndrome following attempted hanging. Rev. Neurol. 1993, 149, 213–216.
- Johkura, K.; Naito, M. Wernicke’s encephalopathy-like lesions in global cerebral hypoxia. J. Clin. Neurosci. 2008, 15, 318–319.
- Molavi, M.; Vann, S.D.; de Vries, L.S.; Groenendaal, F.; Lequin, M. Signal change in the mammillary bodies after perinatal asphyxia. AJNR Am. J. Neuroradiol. 2019, 40, 1829–1834.
- Miller, S.P.; Ramaswamy, V.; Michelson, D.; Barkovich, J.; Holsuser, B.; Wycliffe, N.; Glidden, D.V.; Deming, D.; Partridge, J.C.; Wu, Y.W.; et al. Pattern of brain injury in term neonatal encephalopathy. J. Pediatr. 2005, 146, 453–460.
- De Vries, L.S.; Groenendaal, F. Patterns of neonatal hypoxic-ischaemic brain injury. Neuroradiology 2010, 52, 555–566.
- Parikh, P.; Juul, S.E. Neuroprotective strategies in neonatal brain injury. J. Pediatr. 2018, 192, 22–32.
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R. Soc. B 2005, 360, 2335–2345.
- Jhala, S.S.; Hazel, A.S. Modeling neurodegenerative disease pathophysiology in thiamine deficiency consequences of impaired oxidative metabolism. Neurochem. Int. 2011, 58, 248–260.
- Wassink, G.; Gunn, E.R.; Drury, P.P.; Bennet, L.; Gunn, A.J. The mechanisms and treatment of asphyxia encephalopathy. Front. Neurosci. 2014, 8, 40.
- Johnston, M.V.; Fatemi, A.; Wilson, M.A.; Northington, F. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol. 2011, 10, 372–382.
- Kuan, C.-Y.; Chen, H.-R.; Gao, N.; Kuo, Y.-M.; Chen, C.-W.; Yang, D.; Kinkaid, M.M.; Hu, E.; Sun, Y.-Y. Brain-targeted hypoxia-inducible factor stabilization reduces neonatal hypoxic-ischemic brain injury. Neurobiol. Dis. 2021, 148, 105200.
- Zera, K.; Zastre, J. Thiamine deficiency activates hypoxia inducible factor-1α to facilitate pro-apoptotic responses in mouse primary astrocytes. PLoS ONE 2017, 12, e0186707.
- Chavez, J.C.; Baranova, O.; Lin, J.; Pichiule, P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. J. Neurosci. 2006, 26, 9471–9481.
- Trollmann, R.; Gassmann, M. The role of hypoxia-inducible transcription factors in the hypoxic neonatal brain. Brain Dev. 2009, 31, 503–509.
- Greijer, A.; Van der Wall, E. The role of hypoxia inducible factor 1(HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 100914.
- Liang, X.; Liu, X.; Lu, F.; Zhang, Y.; Jiang, X.; Ferriero, D.M. HIF1α signalling in the endogenous protective responses after neonatal brain hypoxia-ischemia. Dev. Neurosci. 2018, 40, 617–626.
- Davidson, J.O.; Wassink, G.; van den Heuj, L.G.; Bennet, L.; Gunn, A.J. Therapeutic hypothermia for neonatal hypoxic-ischemic encephalopathy: Where to from here? Front. Neurol. 2015, 6, 198.
- Pauliah, S.S.; Shankaran, S.; Wade, A.; Cady, E.B.; Thayyl, S. Therapeutic hypothermia for neonatal encephalopathy in low- and middle-income countries: A systematic review and meta-analysis. PLoS ONE 2013, 8, e58834.
- Ferriero, D.M. Neonatal brain injury. N. Engl. J. Med. 2004, 351, 1985–1995.
- Groenendaal, F.; Lammers, H.; Smit, D.; Nikkels, P.G.J. Nitrotyrosine in brain tissue of neonates after perinatal asphyxia. Arch. Dis. Child. Fetal Neonatal Ed. 2006, 91, F429–F433.
- Okai, Y.; Higashi-Okai, K.; Sato, E.F.; Konaka, R.; Inoue, M. Potent radical-scavenging activities of thiamin and thiamin diphosphate. J. Clin. Biochem. Nutr. 2007, 40, 42–48.
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125.
- Stepuro, A.I.; Piletskaya, T.P.; Stepuro, I.I. Role of thiamine thiol form in nitric oxide metabolism. Biochemistry 2005, 70, 339–349.
- Calingasan, N.Y.; Gibson, G.E. Vascular endothelium in a site of free radical production and inflammation in areas of neuronal loss in thiamine-deficient brain. Ann. N. Y. Acad. Sci. 2000, 903, 353–356.
- Gong, P.; Hua, R.; Zhang, Y.; Zhao, H.; Tang, Z.; Mei, X.; Zhang, M.; Cui, J.; Li, C. Hypothermia-induced neuroprotection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J. Cereb. Blood Flow Metab. 2013, 33, 928–934.
- Menezes, R.R.; Godin, A.M.; Rodrigues, F.F.; Coura, G.M.E.; Melo, I.S.F.; Brito, A.M.S.; Bertollo, C.M.; Paulino, T.P.; Rachid, M.A.; Machado, R.R.; et al. Thiamine and riboflavin inhibit production of cytokines and increase the anti-inflammatory activity of a corticosteroid in a chronic model of inflammation induced by complete Freund’s adjuvant. Pharmacol. Rep. 2017, 69, 1036–1043.
- Gunn, A.J.; Thoresen, M. Hypothermic neuroprotection. NeuroRx 2006, 3, 154–169.
More
Information
Subjects:
Allergy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
31 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No