The precise function of CERKL, a Retinitis Pigmentosa (RP) causative gene, is not yet fully understood. There is evidence that CERKL is involved in the regulation of autophagy, stress granule generation and mitochondrial dynamics, and it is considered a resilience gene that protects retinal cells from oxidative stress. Mutations in most RP genes affect photoreceptors, but retinal pigment epithelium (RPE) cells may be also affected. Upon oxidative stress conditions, CERKL knockdown causes RPE mitochondrial fragmentation and respiratory metabolism alterations, whereas CERKL overexpression protects mitochondria of RPE cells. Particular retinal phenotypic traits observed in patients carrying CERKL mutations may reflect a combination of photoreceptor and RPE alterations.
1. Introduction
Retinitis pigmentosa (RP) comprises a genetically heterogenous group of retinal degenerative diseases characterized by night blindness and progressive loss of vision due to photoreceptor degeneration. To date, more than 70 causative genes have been identified
[1]. Mutations in
CERKL (
CERamide Kinase Like) have been reported to cause non-syndromic autosomal recessive RP
[2] as well as cone-rod dystrophy (CRD)
[3].
CERKL expression is highly complex, with more than 20 transcripts and several alternative promoters in human and mouse tissues
[4][5].
2. KD/KO RPE Displays More Polynucleated Cells and Smaller Mitochondria
CERKL localizes at different cellular compartments, e.g., nuclei, Golgi apparatus, and mitochondria, among others, in both immortalized and primary cell lines
[6][7][8][9]. However, little is known about the subcellular localization of CERKL in mammalian RPE. To assess whether endogenous CERKL colocalizes with mitochondria in murine RPE, we performed immunofluorescence of retinal cryosections from
WT/WT mice with the mitochondrial marker COX-IV and two different in-house antibodies against peptides encoded by exon 2 (anti-CERKL2) or exon 5 (anti-CERKL5), which detect different pools
of endogenous isoforms
[5]. In both cases, a pool of CERKL colocalizes with mitochondria (
Figure 1A).
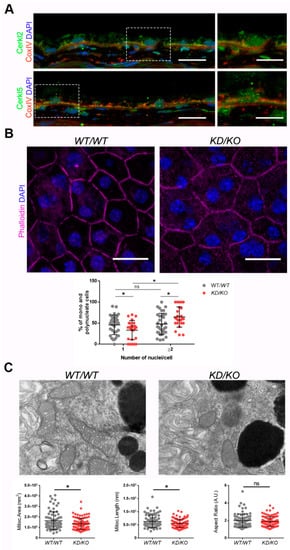
Figure 1. The Retinal Pigment Epithelium (RPE) of KD/KO mice shows morphological alterations.(A) CERKL is expressed in RPE as detected by immunofluorescence of retinal cryosections from WT/WT mice with COX-IV (red) and anti-CERKL2 or anti-CERKL5 (green) antibodies. Regions of interest (ROIs) represent higher magnification from left panels. Scale bar: 20 μm (lower magnification) or 10 μm (higher magnification). (B) The RPE from KD/KO mice shows an increased number of polynucleated cells. RPE flat mounts from WT/WT and KD/KO mice stained with phalloidin (for F-actin, magenta) and 4′,6-diamidino-2-phenylindole (DAPI, blue) and imaged by confocal microscopy were used to quantify the percentage of mono and polynucleated cells. Scale bar: 20 μm. (C) Mitochondrial fragmentation in KD/KO RPE. Transmission electron microscopy (TEM) microphotographies of retinal pigment cells from WT/WT and KD/KO (2-month-old mice) were used to quantify mitochondrial area, length, and aspect ratio. Scale bar: 500 nm. The data are expressed as the mean ± SD, n = 25–34 ROIs from 3 animals per group in (B) and n = 78–89 mitochondria from 3 animals per group in (C). Statistical analysis by Mann–Whitney test: * p-value ≤ 0.05.
Detrimental factors, such as aging or oxidative stress, may compromise RPE homeostasis. Aging and blinding diseases, including rare retinopathies, are associated with changes to RPE structure, such as the increasing of bi- and multinucleated RPE cells
[10][11]. Multinuclear cell formation has been proposed as a mechanism to compensate the apoptotic loss of RPE cells and to maintain the epithelial layered structure under stress conditions
[12]. As
CERKL is proposed as a resilience gene against oxidative stress in mammalian retina, and its depletion in
KD/KO mice causes retinal degeneration, we investigated if
KD/KO RPE suffered changes in the percentage of mono- and multinucleated cells. We observed that the ratio of mono- and poly-nucleated cells is shifted in
KD/KO RPE, with an increase in poly-nucleated cells with a decrease in mono-nucleated cells (
Figure 1B).
The preservation of mitochondrial function and morphology is essential to ensure RPE homeostasis and a correct crosstalk with neuroretina. Since a pool of CERKL colocalizes at mitochondria in both RPE and neuroretina, we assessed possible alterations of mitochondrial morphology in KD/KO RPE in vivo by transmission electron microscopy (TEM) of WT/WT and KD/KO RPE from 2-month-old mice. We found a significant decrease in mitochondrial area and length in KD/KO, without changes in the morphological parameter aspect ratio (Figure 1C). These findings were further confirmed by performing the same morphological analysis of mitochondria in 8-month-old mice. On the other hand, no detectable changes in the size of the Bruch’s membrane, of which the structure is typically altered in several retinopathies, could be observed.
Overall, our data indicated that the depletion of Cerkl expression alters cell structure and mitochondrial morphology in RPE cells in vivo.
3. CERKL-silenced ARPE-19 Cells Show Alterations in Mitochondrial Network
Since depletion of Cerkl in KD/KO mouse model cause alterations in the mitochondrial network of RPE, we studied the deficiency of CERKL in vitro, taking advantage of ARPE-19 cells treated with small interference (si) RNAs against CERKL. To evaluate the effect of siRNAs on CERKL expression, we tested two different control siRNAs (scr1 and scr2) and two siRNAs against different regions of CERKL (siCERKL1 and siCERKL2) on ARPE-19 cells through immunofluorescence using an in-house antibody that recognizes most isoforms of the human CERKL protein (anti-CERKL). Phalloidin was used to delimit cell area (Figure 2A).
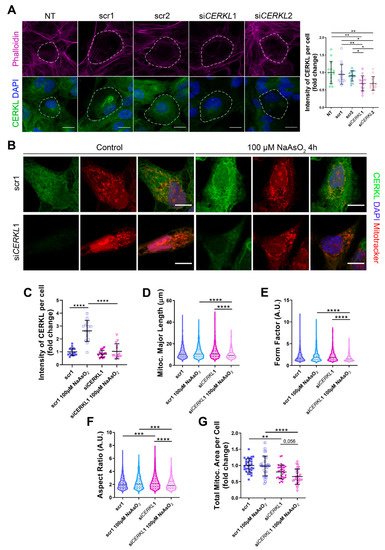
Figure 2. Mitochondrial morphology is altered in CERKL-silenced human ARPE-19 cells. (A) siRNAs against CERKL decrease CERKL expression in ARPE-19 cells. Immunofluorescence confocal images of non-transfected, negative control (scr1 and scr2) and CERKL-silenced (siCERKL1 and siCERKL2) differentiated ARPE-19 cells stained with phalloidin (magenta), CERKL (green), and DAPI (blue) were used to quantify CERKL fluorescence intensity per cell. Scale bar: 15 μm. (B) Mitochondrial fragmentation in CERKL-silenced cells under stress conditions. Immunofluorescence images from a single focal plane of negative control (scr1) and CERKL-silenced (siCERKL1) differentiated ARPE-19 cells in the control and under oxidative stress conditions were used to quantify CERKL fluorescence intensity (C), mitochondrial major length (D), form factor (E), aspect ratio (F), and mitochondrial area (G). Cells were stained with CERKL (green) and DAPI (blue), and mitochondria were marked with Mitotracker (red). Scale bar: 10 μm. Data are represented as the mean ± SD (A,C,G) and as violin plots (D–F), n = 15 cells per condition (A,C) and n = 565–997 mitochondria (D–F) from n = 26–33 cells (G) per condition. Statistical analysis by one-way ANOVA (A,C,G) and Kruskal–Wallis (D–F) tests: * p-value ≤ 0.05, ** p-value ≤ 0.01, *** p-value ≤ 0.001, **** p-value ≤ 0.0001.
Quantification of CERKL fluorescence intensity revealed a significant decrease in CERKL expression in both siCERKL1 and siCERKL2-treated cells. The most significant difference in CERKL expression occurred between scr1 and siCERKL1 siRNAs and were thus selected for subsequent experiments (Figure 2A).
Due to genetic and environmental factors, the retina, including RPE, is constantly under stress conditions that may damage mitochondrial network organization if resilience mechanisms do not work properly
[13]. To assess CERKL function in regulating the morphology of the mitochondrial network in retinal epithelium cells, we knocked-down
CERKL expression in human ARPE-19 cells using si
CERKL1 under basal and oxidative stress conditions (100 μM sodium arsenite for 4 h) and studied mitochondrial morphology through the fluorescent mitochondrial tracker Mitotracker (
Figure 2B). In accordance with previous studies, a pool of CERKL localizes at the mitochondria. Remarkably, CERKL total expression is significantly induced in ARPE-19 cells under oxidative stress conditions. Notably, this sodium arsenite-dependent boost expression of CERKL is completely impaired when
CERKL is downregulated by si
CERKL1 (
Figure 2C).
Analyses of mitochondrial morphology revealed a significant decrease in mitochondrial major length, form factor, and aspect ratio in CERKL-silenced cells compared with control cells under stress conditions. In addition, these morphological parameters were significantly diminished in CERKL-silenced cells when comparing oxidative stress conditions with basal conditions (Figure 2D–F). Moreover, we found a significant increase in aspect ratio between control and CERKL-silenced cells both in basal conditions (Figure 2F). CERKL-silenced cells showed significantly decreased mitochondrial area in basal conditions compared with controls, and this decrease was more evident under stress conditions (Figure 2G).
To sum up, CERKL deficiency alters the mitochondrial network in ARPE-19 cells, particularly in response to oxidative stress conditions.
4. CERKLb and CERKLc Overexpression Restores Mitochondrial Network under Oxidative Stress in ARPE-19 Cells
CERKL overexpression has been described as protective against oxidative stress by interacting with antioxidant enzymes
[6], regulating autophagy
[7], and downregulating apoptosis
[14], among others. In the human retina, due to different alternative splicing events,
CERKL produces at least four different protein isoforms (CERKLa, CERKLb, CERKLc, and CERKLd)
[14] that display different protein domains.
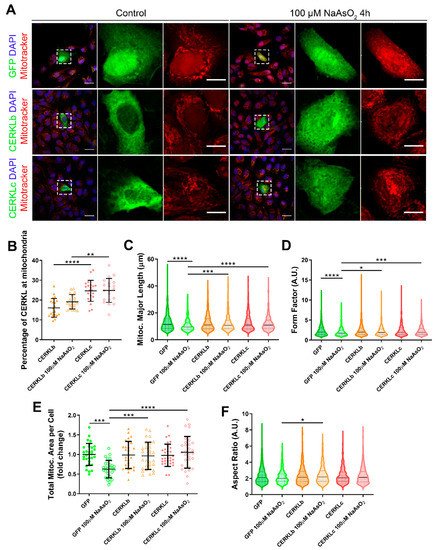
In order to test if different isoforms may differentially regulate mitochondrial dynamics, we overexpressed human CERKLb and CERKLc isoforms in ARPE-19 cells. The CERKLb isoform is the longest one (558 aa) and includes a human-specific additional exon (4b), whereas CERKLc is the shortest isoform (419 aa) and lacks the ATP-binding site and the diacylglycerol kinase (DAGK) domain. The two isoforms display a pleckstrin-homology domain (including a mRNA binding domain) and the nuclear localization and export signals. ARPE-19 cells transfected with either CERKLb or CERKLc isoforms were treated with 100 μM sodium arsenite for 4 h, and the morphology of their mitochondrial network was assessed by immunofluorescence using Mitotracker (Figure 3A). CERKLb localizes more diffusely within the cell, including the nucleus, whereas CERKLc is 10% more concentrated in mitochondria under basal and oxidative stress conditions (Figure 3A,B).
Figure 3. Mitochondrial morphology is restored in ARPE-19 cells under oxidative stress conditions when CERKL isoforms are overexpressed. (A) CERKL overexpression protects the mitochondrial network under oxidative stress conditions. Immunofluorescence images from a single focal plane of differentiated ARPE-19 cells transfected with pEGFP-N2, CERKLb-GFP, and CERKLc-GFP in the control and under oxidative stress conditions were used to quantify the percentage of CERKL at mitochondria (B), mitochondrial major length (C), form factor (D), mitochondrial area (E), and aspect ratio (F). Mitochondria were marked with Mitotracker (red), and nuclei were counterstained with DAPI (blue). Scale bars: 25 μm and 10 μm in higher magnification images. Data are represented as the mean ± SD (B,F) and as violin plots (C–E), n = 20–21 cells (B) and n = 404–726 mitochondria (C–E) from n = 28–30 cells (F) per condition. Statistical analysis by one-way ANOVA (B,F) and Kruskal–Wallis (C–E) tests: * p-value ≤ 0.05, ** p-value ≤ 0.01, *** p-value ≤ 0.001, and **** p-value ≤ 0.0001.
Morphological analyses showed a statistically significant decrease in mitochondrial major length, area, and form factor in GFP-transfected cells (controls) under oxidative stress compared with in the basal condition. However, mitochondria in cells transfected with both CERKLb and CERKLc did not show any significant change in these parameters between basal and oxidative stress conditions. In fact, the mitochondrial length, area, and form factor in CERKLb and CERKLc-transfected cells under stress conditions were not affected in contrast with the decrease in all these measurements in stressed control cells (Figure 3C–F).
Altogether, these results strongly indicated that both CERKLb and CERKLc localized at mitochondria and that CERKL overexpression in ARPE-19 cells protected mitochondrial network under oxidative stress conditions.
5. Mitochondrial Superoxide Production Is Reduced by CERKLb and CERKLc Overexpression in ARPE-19 Cells
Mitochondrial network disorganization is a typical sign of cellular damage and loss of mitochondrial homeostasis
[15]. Under oxidative stress or pathogenic conditions, damaged mitochondria generate free radicals, such as mitochondrial superoxide, due to impaired antioxidant response
[16]. Therefore, we measured mitochondrial oxidation using MitoSOX quantification in cells either overexpressing CERKLb and CERKLc or depleted in
CERKL expression under both control and oxidative stress conditions (
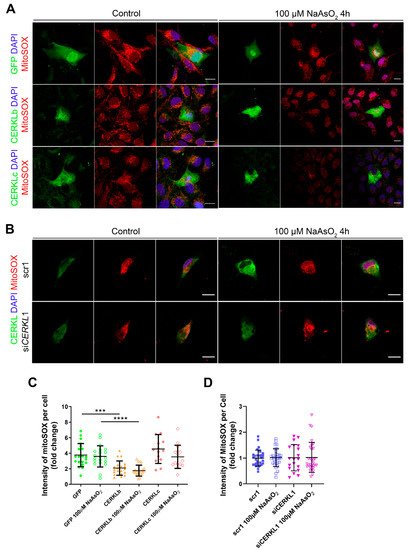
Figure 4A,B). A significant decrease in MitoSOX intensity was observed when overexpressing CERKLb and CERKLc in basal conditions, and this mitochondrial protection was maintained under oxidative stress (
Figure 4C). On the other hand, neither control cells nor cells depleted in
CERKL showed an increase in MitoSOX in any condition, thus pointing to other factors being necessary for mitochondrial superoxide production besides stress by sodium arsenite (
Figure 4D).
Figure 4. Quantification of MitoSOX in ARPE-19 cells overexpressing CERKLb and CERKLc or knocking-down CERKL. (A) Overexpression of CERKLb and CERKLc reduces the production of mitochondrial superoxide. Immunofluorescence images from a single focal plane of differentiated ARPE-19 cells transfected with pEGFP-N2, CERKLb-GFP, and CERKLc-GFP under control and stress conditions were used to quantify MitoSOX fluorescence intensity (C). Mitochondrial superoxide was detected with MitoSOX (red), and nuclei were counterstained with DAPI (blue). Scale bar: 15 μm. (B) CERKL depletion is not sufficient to increase mitochondrial superoxide production under basal and stress conditions. Immunofluorescence images from a single focal plane of negative control (scr1) and CERKL-silenced (siCERKL1) differentiated ARPE-19 cells under basal and oxidative stress conditions were used to quantify MitoSOX fluorescence (D). Cells were stained with CERKL (green) and DAPI (blue), and mitochondrial superoxide was detected with MitoSOX (red). Scale bar: 15 μm. Data are represented as the mean ± SD, n = 28–33 cells per condition. Statistical analysis by Kruskal–Wallis test: *** p-value ≤ 0.001 and **** p-value ≤ 0.0001.
In summary, these findings pointed to a protection of mitochondria as measured by mitochondrial superoxide production when CERKLb and CERKLc were overexpressed.
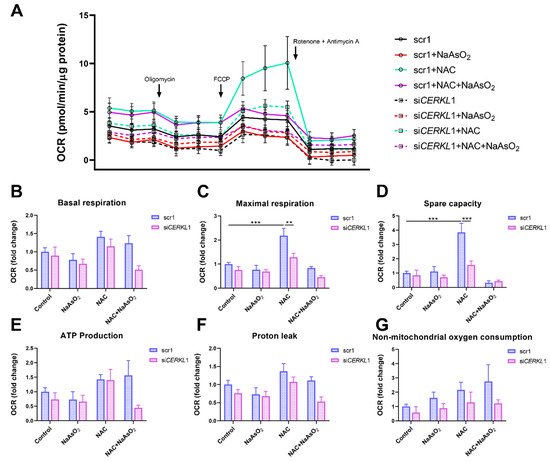
6. CERKL Knock-Down Alters Mitochondrial Respiration
Considering that (i) CERKL deficiency affects mitochondrial network organization in ARPE-19 cells, and (ii) oxygen consumption and other mitochondrial energy-related measurements in neural retinas of KD/KO mice are compromised, we performed Seahorse analysis to evaluate mitochondrial function and oxygen consumption rate (OCR) in CERKL-depleted ARPE-19 cells. We compared mitochondrial performance in control and stress conditions as well as under antioxidant pre-treatment (4 μM N-acetyl-L-cysteine (NAC) for 24 h).
Differentiated ARPE-19 cells displayed low oxygen consumption rates, which made changes between conditions very subtle. Under control conditions, OCR was reduced (black straight versus dotted lines) upon knock-down of CERKL expression (Figure 5A). In fact, all respiration-related parameters, including maximal respiration and non-mitochondrial oxygen consumption, showed a clear trend of diminishing in CERKL-deficient cells (Figure 5B–G). As expected, we found a decrease in the respiration parameters (basal and maximal respiration) after NaAsO2 treatment in both scr1 and siCERKL1-transfected cells, conserving the tendency.
Figure 5. Oxygen consumption is impaired in CERKL-depleted ARPE-19 cells under different antioxidant (NAC pre-treatment) and oxidative stress (NaAsO2 treatment) conditions. (A) Altered oxygen consumption rate (OCR) in CERKL-knocked-down cells in all conditions; straight lines indicate values from scrambled control cells, whereas dotted lines indicate siCERKL1-treated cells. (B) Basal respiration are the initial OCR levels without treatment. (C) Maximal respiration is obtained after the addition of FCCP (carbonyl cyanide-4-trifluoromethoxy phenyl-hydrazone). (D) Spare capacity is calculated by subtracting basal respiration from maximal respiration. Note that CERKL-depleted cells neither increase the respiratory capacity after antioxidant treatment nor maintain maximal respiration and spare capacity after oxidative injury. Oligomycin addition reveals (E) ATP production and (F) proton leak levels. (G) Non-mitochondrial oxygen consumption values are obtained after rotenone and antimycin A addition. Statistical test by two-way ANOVA: ** p-value ≤ 0.01, *** p-value ≤ 0.001.
NAC pre-treatment results in an increase (two-fold) in mitochondrial and non-mitochondrial respiration in control cells, which allows them to resist oxidative stress conditions. However, CERKL-depleted cells are not able to respond to the antioxidant pre-treatment to these high levels and are thus not resilient to oxidative injury. Remarkably, mitochondrial respiration is decreased in CERKL-depleted cells, while non-mitochondrial oxygen consumption increases slightly in all conditions, probably as a cell compensatory mechanism.
Overall, these results denoted that CERKL-deficient ARPE-19 cells do not respond to antioxidant (NAC) protective pre-treatment and are thus not as resilient to oxidative stress as control cells.
7. Conclusions
Our results further reinforce the role of CERKL as a resilience gene. On the one hand, oxidative stress treatment induces the expression of CERKL as a very early cell response: we observed a more than two-fold increase in endogenous CERKL 4 h after the addition of the arsenite reagent. On the other hand, overexpression and depletion of CERKL expression cause opposite effects on mitochondrial shape and size in RPE cells. Under oxidative stress, CERKL overexpression protects RPE mitochondrial morphology, whereas depletion in CERKL causes mitochondrial fragmentation, respiratory alterations, and polynucleation (a signal of dysfunction and stress) in RPE.
In conclusion, this work supports that one of the physiological roles of CERKL is to protect the mitochondrial network in retinal cells, both neurons and epithelium, against the injury of light/oxidative stress. Mutations in many RP genes, such as CERKL, can potentially affect both tissues. Therefore, some unusual and particular phenotypic traits of the RPE observed in the retinas of patients carrying CERKL mutations may reflect the combination of photoreceptor and RPE alterations. We propose that by identifying RPE pathogenic events, particularly those related to oxidative stress, we could design specific drug- or cell-based therapies that target RPE to improve photoreceptor homeostasis and/or to halt photoreceptor neurodegeneration.
 +1 credit
+1 credit