 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Valentijn De Coster | + 17925 word(s) | 17925 | 2020-08-18 10:19:16 | | | |
| 2 | Felix Wu | -6451 word(s) | 11474 | 2020-11-21 13:33:06 | | |
Video Upload Options
Supported nanoparticles are commonly applied in heterogeneous catalysis. The catalytic performance of these solid catalysts is, for a given support, dependent on the nanoparticle size, shape, and composition, thus necessitating synthesis techniques that allow for preparing these materials with fine control over those properties. Such control can be exploited to deconvolute their effects on the catalyst’s performance, which is the basis for knowledge-driven catalyst design. In this regard, bottom-up synthesis procedures based on colloidal chemistry have proven successful in achieving the desired level of control for a variety of fundamental studies. This article aims to give an overview of recent progress made in this synthesis technique for the application of controlled catalytic materials in gas-phase catalysis. The focus goes to mono- and bimetallic materials, as well as to recent efforts in enhancing their performance by embedding colloidal templates in porous oxide phases.
1. Introduction
With an approximate contribution of 80 – 90 % to all chemical processes [1], heterogeneous catalysis, wherein a gas- or liquid-phase reaction takes place over a solid catalyst, is indispensable in today’s society. Supported nanoparticles (NPs) – which can be either metals, metal oxides or sulfides – are particularly attractive in this discipline, as they combine the high activity related to the catalytically active sites on the NP surface with the inherent thermal stability of support materials [2][3][4]. More so, interactions at the metal-support interface, called metal-support interactions (MSI), can significantly alter the catalytic behavior of the NPs, further proving the significance of these material combinations [5][6][7].
For a given metal and support, the catalytic activity, which is commonly expressed as turnover frequency (TOF), product selectivity and stability are dependent on the active sites exposed on the NP surface. In turn, these are a function of the NP size, shape and composition [8][9][10][11][12][13]. To advance catalyst development, it is primordial to unambiguously establish correlations between these structural parameters and the catalyst’s performance. Such property-performance relationship studies help identify those catalyst properties that are responsible for enhanced catalyst performance, which, in turn, forms the basis of a knowledge-driven design of novel improved catalysts. However, such an approach requires model catalysts prepared by synthesis strategies that allow independent control over the structural parameters.
While single crystals have well-defined surface characteristics, these model materials are far from the more complex supported NPs that make up industrially relevant solid catalysts, thus making them unfit for investigating the many factors affecting catalytic behavior [14][15][16]. Moreover, single crystal studies are mostly performed under ultra-high vacuum (UHV) conditions [14]. This is in strong contrast with true reactive conditions which are closer to ambient pressures. These differences between model versus true materials and UHV versus elevated pressure testing conditions are respectively denoted as the “material gap” and “pressure gap” [16][17]. To cross these gaps, the synthesis of well-defined supported NPs is required. However, traditional wet chemical catalyst synthesis methods for the creation of such materials, such as wet impregnation (WI) [3][18], (co-)precipitation [3][19], ion exchange [18] or deposition-precipitation (DP) [20], mostly result in ill-defined property distributions, making them unsuitable for controlled catalyst synthesis.
To achieve the desired level of control, the application of colloidal NPs has proved an efficient strategy. Recent developments in wet chemical colloidal synthesis protocols have resulted in a high level of control over the size, shape and composition of colloidal NPs [11][21][22][23]. As such, the use of metal colloids as a component of catalyst design, e.g. by the deposition of separately synthesized colloids onto a support, has gained increased attention for the controlled creation of catalysts with the appropriate level of complexity. Furthermore, recent developments have led to protocols for the creation of “embedded” NPs wherein the colloidally prepared NPs are enveloped by the support rather than supported onto it, which has opened a novel chapter for highly stable catalysts [4][24][25][26][27].
This review covers recent progress in the application of catalysts with uniform properties prepared by colloidal synthesis. General principles of the synthesis procedures are first elaborated, followed by catalytic applications of mono- and bimetallic supported NPs prepared via colloidal techniques. In view of the importance of improving catalyst stability, applications of catalysts with oxide-coated NPs are discussed. Finally, concluding remarks are given regarding the state-of-the-art of catalytic applications of these synthesis techniques, along with an outlook on their utilization and development in future studies.
2. Colloidal Synthesis in Gas-Phase Catalysis
2.1. Principles of Colloidal Synthesis for Supported Catalyst Preparation
By definition, a metal colloid, also called a “sol”, denotes a suspension of metal particles, typically in the range of 1 – 1000 nm, suspended in a liquid medium [28][29]. Herein, the particulates are covered by a protective layer that prevents them from coalescing into larger aggregates. The use of colloids is one of the most versatile methods in tuning catalyst properties, as it allows the creation of metallic, i.e. zerovalent, NPs independently of the support. Hence, this synthesis method allows for great tunability of the catalyst’s properties, provided the NPs are created in a controlled manner.
Methods for creating colloidal metals are classified in two categories: “top-down” or “bottom-up” [30]. Top-down refers to physical approaches that create NPs from the dispersion of bulk materials into their NP building blocks. Examples include, but are not limited to, laser ablation [31], chemical etching [32] and mechanical milling [33]. While such approaches usually produce relatively large quantities of NPs (~g), this advantage is countered by their broad particle size distributions, low synthesis reproducibility and the relative complexity of the instrumentation involved [21]. Bottom-up approaches, on the other hand, entail chemical routes whereby atoms or molecules are assembled into larger nanoscale (1 – 100 nm) structures. For the purpose of catalyst synthesis, the most common approaches are chemical reduction of metal precursors, i.e. where an external reagent is used to establish the reduction, and decomposition of organometallic precursors via thermal, photochemical, sonochemical or radiolytic means [34], among which thermal decomposition is the most established [29]. While bottom-up methods yield smaller quantities of NPs (~mg) than top-down, they have the benefit of allowing better control over the NP morphology, a higher level of synthesis reproducibility as well as requiring less sophisticated equipment [21][21]. The aforementioned factors motivate the use of bottom-up approaches for colloidal synthesis of well-controlled supported NPs. Consequently, only these will be covered in this review.
Bottom-up colloidal synthesis relies on the controlled natural growth of suspended NPs. In a typical chemical reduction-based synthesis (Figure 1a), this entails the dissolution of precursors of the desired metal into a solvent in the presence of a protective agent. In the following step, the reduction process proceeds at elevated temperature to generate metallic NPs by introducing a reducing agent. In the case of protocols based on thermal decomposition, two common approaches are applied. The first, represented in Figure 1b, is near-identical to the chemical reduction approach described above; it only differs in the fact that no reducing agent is applied at elevated temperature and that this temperature is mostly higher than in the case of chemical reduction methods, e.g. ~200 °C versus ~100 °C. Another protocol relies on “hot injection” of the metal precursor (Figure 1c). Herein, solely the solvent and protective agent are first added to a reaction vessel. After heating this mixture, the precursor – typically dissolved in the same solvent as present in the reaction vessel – is injected into the ‘hot’ reaction mixture. After a certain dwell time at the final temperature, the synthesis is terminated by cooling off the system.
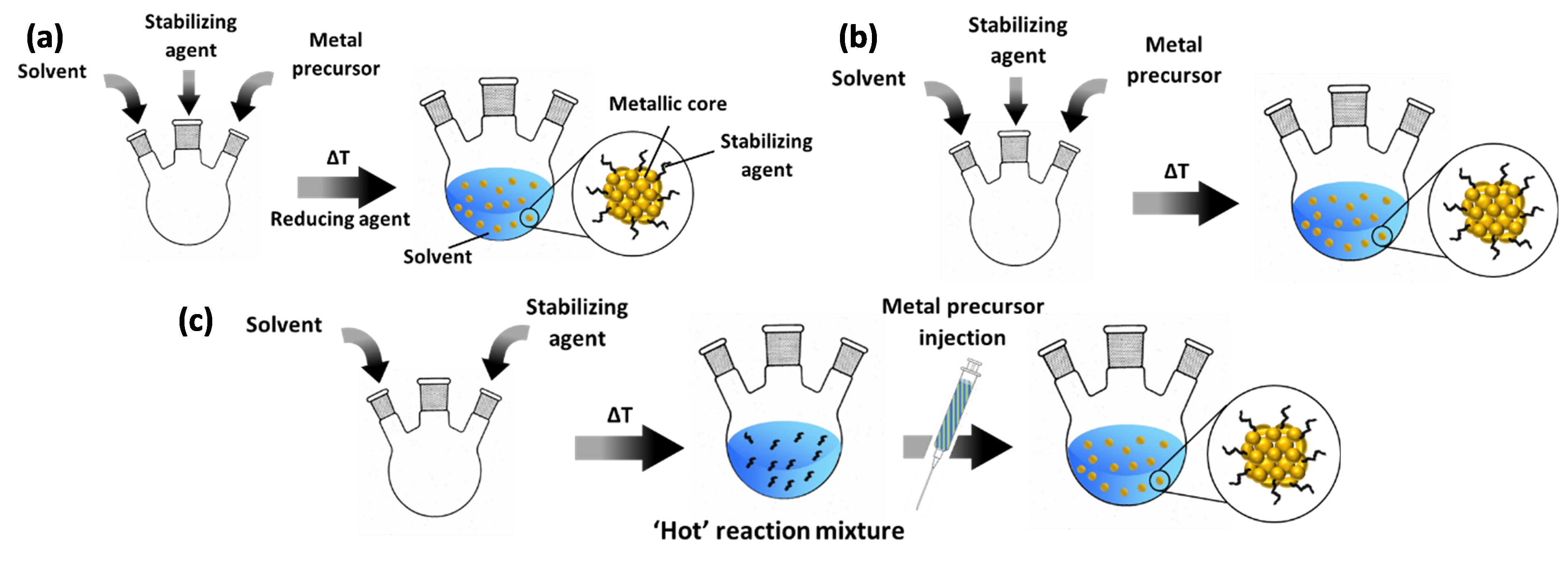
Figure 1. Common protocols for colloid synthesis: (a) chemical reduction; (b) thermal decomposition with precursor added in the initial step; (c) thermal decomposition via hot injection. The blue-green content of the syringe indicates that the metal precursor (green) is dissolved in the same solvent as the reaction vessel (blue). For ease of representation, the final step in the synthesis, i.e. cooling down of the reaction vessel, is not included.
From the above, it follows that bottom-up synthesis protocols comprise three key components: a metal precursor, a solvent and a protective agent [13]. In the case of chemical reduction protocols, an external reducing agent is distinguished as an additional fourth component. It should be noted that these components are not necessarily physically separate; a certain chemical may act as more than one component. An example thereof are alcohols, which can act as solvent, protective agent and reducing agent [35][36].
The most common metal precursors are ionic salts, examples of which are nitrates, chlorides, sulfates, acetates and acetylacetonates [13]. However, organometallic complexes are an equally viable option. Solvents provide the continuous liquid phase of the colloid and are either aqueous or organic. Protective agents envelop the particulates through interaction (e.g. adsorption or coordination), forming a protective shell with a twofold purpose [37]. First, it regulates the growth of the preformed NPs by controlled diffusion of precursor from the surrounding liquid phase through this shell into the colloid’s core. Moreover, this protective layer prevents NP agglomeration through its stabilizing role, which is steric, electrostatic or electrosteric in nature [28][34]. Due to the growth-controlling and stabilizing roles of protective agents, they are also called capping or stabilizing agents. A diverse range of such agents has been used in colloidal synthesis, common ones being linear (e.g. polyvinylpyrrolidone (PVP) [38]) or dendrimeric polymers (e.g. polyamidoamine (PAMAM) [39]), which adsorb onto the NP surface and impede coalescence through steric interaction with similarly-protected NPs. Another frequently-used option is ligand-stabilization, referring to the use of molecules with functional groups (e.g. thiols [40], amines [41], organic acids [42] and phosphines [43]), wherein the heteroatom coordinates with the metallic atoms of the NP, thus providing steric stabilization. Microemulsions [44] or reverse micelles [45] through the use of surfactants are also widely applied. In view of chemical reduction, common reducing agents include [34]: alcohols (e.g. glycerol), gases (e.g. H2), hydrides (e.g. NaBH4), superhydrides (e.g. LiBEt3H), amine borane complexes (e.g. borane tert-butylamine complex (BTB)) and hydrazine.
The monodispersity, i.e. the uniformity in NP properties, inherent to bottom-up approaches arises from the separation of a short burst nucleation, wherein many nuclei are created at the same time, and the subsequent growth of these nuclei into NPs without additional nucleation events [13][46]. This can be achieved by a stepwise change in the synthesis environment, such as the addition of a reagent to the synthesis vessel. For chemical reduction methods, it is proposed that the injection of an external reducing agent into a mixture of precursor, solvent and protective agent leads to the formation of many zerovalent nuclei in a short time [47][48]. The incorporation of remaining precursor from the surrounding liquid phase into these nuclei results into precursor reduction and the growth of these NPs. For thermal decomposition of organometallic salts, this separation of nucleation and growth is implemented by either quickly injecting the precursor into a hot (~200 °C) solution containing the capping agent or by imposing a controlled heating ramp to a solution that already contains the capping agent and precursor [49]. The decomposition of said precursors leads to an oversaturation of the solution, which is relieved by nucleation of metallic NPs. This is accompanied by the formation of decomposition products such as CO2 or H2O. As this initial nucleation event decreases the precursor concentration below the critical concentration for nucleation, remaining precursor material can only be further assimilated into the existing nuclei, thus preventing additional nucleation events.
In either method, the protective agent in the reaction mixture ensures the controlled growth of the NPs after the short nucleation event, thus further assuring NP monodispersity. The interaction of the protective agent with the metallic core determines the strength of stabilization by the protective shell. Molecules that are larger (‘bulkier’) and bind more strongly with the metallic core provide greater steric hinderance, slowing down the rate of material addition and decreasing NP size. In combination with the protective agent, the solvent is indispensable in tailoring the colloid’s stability since a favorable interaction between these two components provides an extra barrier to counter NP coalescence.
The precursor material determines the underlying chemistry in the synthesis, such that changing the type of precursor may change the overall NP properties. However, the size, shape and composition of the NPs can be adapted by adjusting several experimental parameters [13][28][49]: the applied temperature; time; the type of protective agent, reducing agent, precursor and solvent used; and the concentrations – or relative molar ratios – of the aforementioned components. Increasing the synthesis temperature promotes the kinetically-driven incorporation of precursor material into the NPs as well as thermodynamically-driven particle growth by Ostwald ripening. Increasing the time spent at elevated temperatures leads to more precursor material being deposited into the NPs, and hence also results in larger NP formation. Upon reaching a set time or temperature, the synthesis vessel is cooled down by removing the heating source or by placing it in a cooling bath, stopping the growth. Increasing the molar ratios of protective agent/precursor favors the creation of more small nuclei, decreasing the NP size. The same effect can be attained through the use of stronger reducing agents. Moreover, protective agents can tailor the NP shape (e.g. spherical, cubic, tetrahedral) by influencing the growth direction through selectively adsorbing onto specific facets of a NP. NP composition is a property exclusive to bi- or multimetallic NPs. Tailoring the composition, i.e. the relative ratios of the elements making up the NP, can be achieved by varying the ratio of precursors in the synthesis or the order of addition to the synthesis vessel, e.g. simultaneously (co-reduction) or sequentially (successive reduction).
To create supported catalysts, colloids can be deposited onto the support by various methods [34][35]. The two most common ones are elaborated here and illustrated in Figure 2. In a first (Figure 2a), pre-synthesized colloids are impregnated onto the support. To this end, the as-prepared colloids can be used. However, since the NP concentration in these mixtures is very low, diffusion-driven migration of the NPs onto the support may take a long time. Adsorption can be stimulated by tuning the pH to promote electrostatic interaction between the NPs and the support [50]. Sonication can also stimulate NP immobilization on the support surface. This relies on the fact that shock waves, which are generated by the collapse of cavitation bubbles, push the NPs towards the support surface with high velocities and thus stimulate NP collision with the support [51][52]. Another option is an intermediary treatment, whereby the colloid is first destabilized through addition of an anti-solvent, resulting in NP flocculation, and subsequent centrifugation [53]. Contrary to the use of as-prepared colloids, this allows for “size-selective precipitation” [54] as it stimulates the precipitation of larger NPs, while smaller ones remain within the supernatant. The selected phase can then be dispersed in an anti-solvent followed by repeated centrifugation to fine-tune the particle size distribution. The final selection of NPs is dispersed in a solvent and impregnated onto the support.
The second method (Figure 2b), denoted as “in situ reduction”, involves the reduction of metal precursors in a mixture that already contains the support, which eliminates the need for a deposition step. In this case, the burst nucleation event is either heterogeneous in nature, i.e. occurring on the support, or forms metallic nuclei in solution, followed by their migration onto the support.
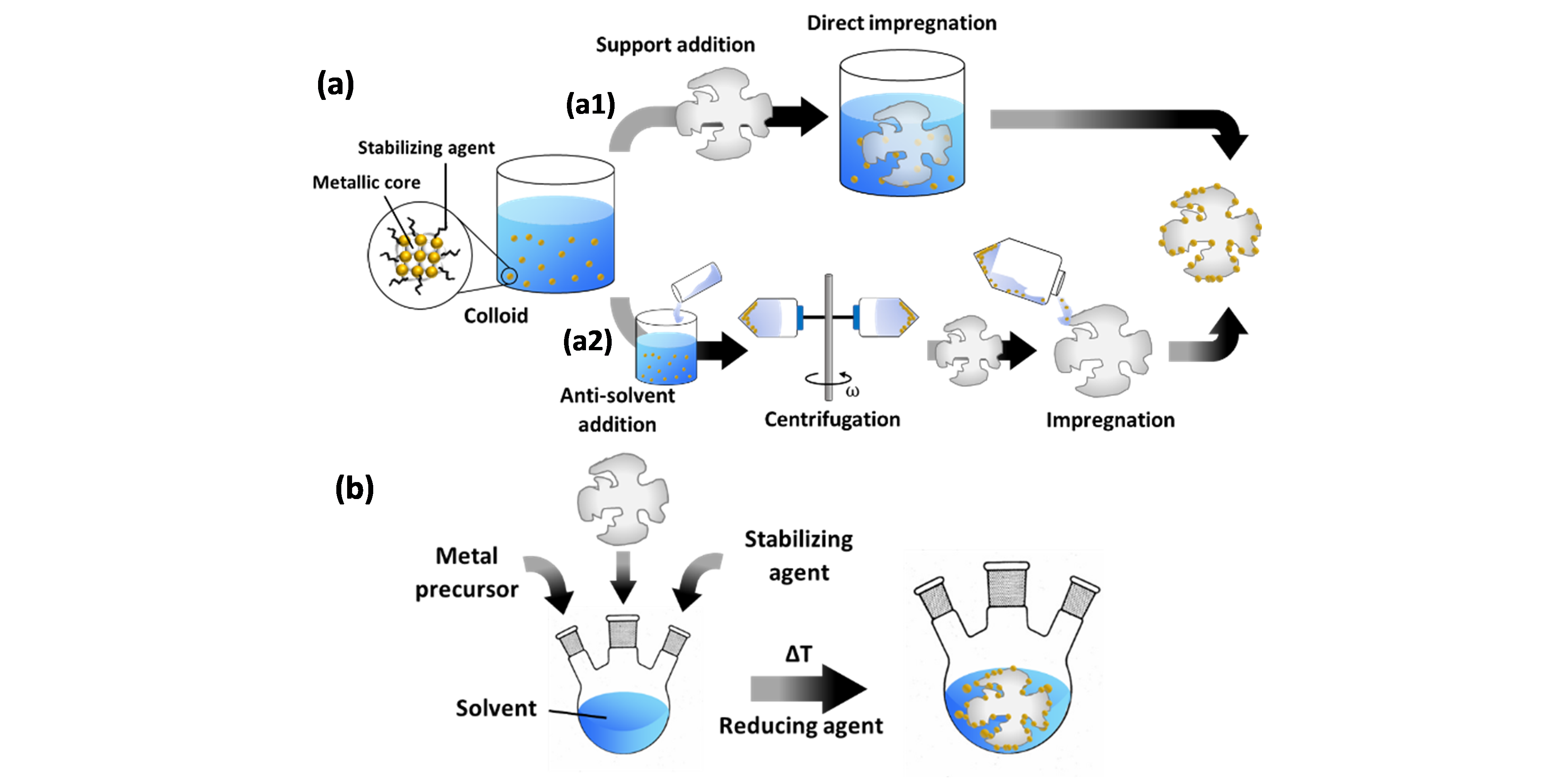
Figure 2. Common methods for supported catalyst preparation via colloidal synthesis. (a) Impregnation methods: (a1) direct impregnation by support addition to the prepared colloid; (a2) an anti-solvent is first added to the colloid, which stimulates NP flocculation, followed by centrifugation of the mixture (with angular velocity ω), and impregnation of the support with the centrifuged supernatant or precipitate phase. (b) In situ reduction method.
Colloids can also be immobilized by grafting them onto the support [34]. This entails functionalizing the support such that the created groups either form chemical bonds with the protective shell or coordinate with the metallic core. While this yields strong immobilization, these approaches have the drawback that the organic-based chemistry requires complex modification steps and that the typically-used sulfur-containing modification compounds can poison the catalyst [55][56].
The spatial distribution of the deposited NPs within the support depends on various factors. In the case of methods which rely on pre-synthesized NPs, the average NP size and support pore size are important parameters; the synthesized NPs must be smaller than the pores in order not to block diffusion-driven transport of the colloid into the support’s pores. If this is not the case, only the exterior surface of the support will be enriched in nanoparticulate material. Should the aforementioned requirements be met, it might still be necessary to wait a long time for the NPs to migrate into the pores as the diffusion of NPs is relatively slow [3][57]. This can be alleviated through sonication of the colloid-support mixture. Herein, the sonication-induced cavitational shock waves not only stimulate NP immobilization on the support surface, but also prevent NPs from blocking the pore openings [57]. In combination with the enhanced mass transport originating from these cavitation phenomena, this allows homogeneous dispersion of the NPs throughout the support channels – provided appropriate sonication times (~h) are applied [57]. In the case of in situ synthesis, the small size of precursor molecules (relative to the NPs formed from them), makes pore size not as stringent as in the case of methods relying on pre-formed NPs. Still, the distribution of the dissolved metal precursors within the support pores is a determining factor for the final spatial distribution of the NPs. Assuming the precursor is well-dissolved in the applied solvent, it follows that the extent of infiltration of the liquid into the pores is a critical factor in achieving homogeneous metal distributions. However, it has been proved that wetting liquids can infiltrate millimeters of a porous body in a matter of seconds or minutes [58]. Hence, attaining well-dispersed metal distributions through in situ synthesis can be considered less problematic than in the case of pre-synthesized NPs. A detailed description on the factors affecting NP and metal precursor spatial distribution is beyond the scope of this work. For more information, reference is made to dedicated literature [3].
In a final preparation step, supports with deposited NPs are separated (e.g. by filtration), dried (e.g. in air, in vacuo or freeze-dried) to remove remaining solvent and finally subjected to a thermal treatment. This treatment aims to remove residual stabilizing agent poisoning the active sites and to bring the catalyst into its active state, which can be either a zerovalent metal, a metal oxide or -sulfide [38][59][60][61][62][63]. However, care must be taken in choosing the conditions, particularly the temperature, of this ‘activation’ as it can induce redistribution of the NPs within the support, as well as phase segregation and sintering, which affect the NP morphology and, in turn, the catalytic performance [3][62][63].
2.2. Monometallic Supported Catalysts by Colloidal Synthesis
Significant progress has been made in the synthesis of monometallic colloids, such that it is now possible to create NPs of any element—provided an appropriate precursor exists—with tunable size and shape [4][21][46]. Specifically, in the field of heterogeneous catalysis, this has allowed for investigating the effect of the aforementioned properties on catalyst performance for a wide range of gas-phase reactions.
2.2.1. Oxidation Reactions
Low-temperature CO oxidation to CO2 is a catalytic reaction that is considered a “probe” or model reaction for other oxidation reactions and has essential applications in emission abatement [64]. In the near-50 years that this reaction has been studied, supported noble metals are among the most efficient reported catalysts [65]. For Pt catalysts, it has been established that interactions of this metal with the support can significantly alter the metal’s electronic properties and, consequently, the catalytic properties. To deconvolute the effects of MSI from NP size effects on the performance of the catalyst, the use of colloidal synthesis as a means to create monodisperse NP size distributions presents an opportunity. In recent work, Xi et al. [66] applied this strategy to study the support effect for Pt/CeO2 and Pt/SiC in CO oxidation. Materials were prepared by support impregnation with Pt NPs of 5.1 nm average size and subsequent calcination in air at 200, 400, 600, or 800 °C for 3 h. For higher calcination temperatures, as-prepared Pt/SiC displayed significant NP agglomeration, while this occurred to a much lesser extent for Pt/CeO2 samples. This was attributed to MSI through the formation of Pt-O-Ce bonds, as indicated by H2 temperature-programmed reduction (H2-TPR) and X-ray photoelectron spectroscopy (XPS). Similar bonds were not found for the SiC samples. In oxidative reaction studies, these interactions had a dual effect. For Pt/CeO2 calcined at 200 or 400 °C, the bonds were weaker, such that these provided active oxygen for CO oxidation without ‘eliminating’ Pt0, which is the active site for CO adsorption. For higher calcination temperatures, the Pt-O-Ce bond became stronger, resulting in the absence of Pt0, thereby deactivating the catalyst and yielding lower TOF values than Pt/CeO2 treated at lower temperatures. As no significant interactions between Pt and SiC were discerned, Pt/SiC calcined at 200 °C yielded lower activities compared to Pt/CeO2 that was treated similarly. However, after calcination up to 800 °C, Pt0 remained abundant in Pt/SiC, as opposed to Pt/CeO2, resulting in higher TOF values for the former catalyst.
In the same context of oxidation reactions, the selective catalytic oxidation of alcohols to aldehydes, ketones and organic acids is an essential reaction in the creation of products with wide applications in the fine-chemicals sector [67]. For the purpose of selective benzyl alcohol oxidation to benzaldehyde, Kumar and co-workers [68] prepared 1 wt% Au/SBA-15 nanocatalysts through colloidal synthesis, as well as through three other synthesis methods, namely homogeneous deposition-precipitation (HDP), microemulsion, and WI, to investigate the effect of the preparation method on the NP dispersion and the catalytic performance. Specifically, for the colloidal method, supported Au NPs were created in situ via a polyol method, i.e., through the use of polyvalent alcohols (polyols). X-ray diffraction (XRD), H2-TPR and transmission electron microscopy (TEM) revealed that the synthesis method increased the NP size in the order HDP < ME < WI < colloidal synthesis, and that the NP dispersion increased in the reverse order. These parameters were correlated with the catalytic performance: HDP-prepared Au/SBA-15 yielded the highest benzyl alcohol conversions and benzaldehyde selectivities of all samples in the temperature range 280–360 °C. Even after regeneration, HDP catalysts still displayed the highest activity of all samples, though the activity was lower compared to the fresh catalyst. This was attributed to a decrease in the number of active sites of Au caused by NP agglomeration, which corroborated TEM images of the spent catalysts.
2.2.2. Hydrogenation Reactions
A comparative study of synthesis methods was also performed by Saadatjou et al. [69] for Rh/γ-Al2O3 with respect to ammonia synthesis. Catalysts were prepared by incipient wetness impregnation (IWI) and an in-situ polyol reduction method. Notable is that the NP size of the IWI catalysts was larger (~12 nm) and more irregular than that of the polyol-synthesized variant (~7 nm). Subsequent reduction and activity measurements yielded the highest activities for the polyol samples. Accounting for the structure-dependency of NH3 synthesis on Ru, this was attributed to the smaller NP size and lower degree of contamination of the metal by aluminum from the support and precursor-related chloride.
In recent years, the reverse reaction of NH3 synthesis, i.e., NH3 decomposition into N2 and H2, has become a topic of interest for COx-free H2 fuel cells for clean energy [70][71][72]. Li and co-workers [73] confirmed the size-dependency of NH3 decomposition on Ni/MCF-17 through the use of uniform NPs with size distributions from 1.5 to 8 nm, tailored by adjustment of the colloid synthesis temperature. A volcano-type relationship was observed between the activity and the Ni NP size with a maximum at ~3 nm for reaction temperatures in the range 550–600 °C (Figure 3a). Theoretical calculations correlated this behavior with a maximum concentration of B5 sites, consisting of five atoms exposing a three-fold hollow hexagonally close-packed site and a bridge site close together, at ~2.5–3 nm (Figure 3b). Such B5 sites have been denoted in literature as the active sites in Ni-based NH3 decomposition catalysts [72][74][75].
Apart from the hydrogenation of N2 to NH3, Fischer-Tropsch synthesis (FTS), i.e., the conversion of a mixture of CO and H2 (syngas) into ‘long’ carbon-chain products, is one of the longest-studied reactions in chemical industry [76][77]. In view of replacing fossil resources with alternative, renewable feedstocks, this reaction has regained interest as it enables the conversion of syngas derived from these alternative resources into clean fuel [78][79]. However, challenges in the design of FTS catalysts remain, more specifically, in the creation of materials that are active, stable, and highly selective towards products with a narrow chain length distribution. As the structure-dependency of FTS is established, the level of control inherent to colloidal synthesis presents an opportunity for fundamental studies and, in turn, rational FTS catalyst design.
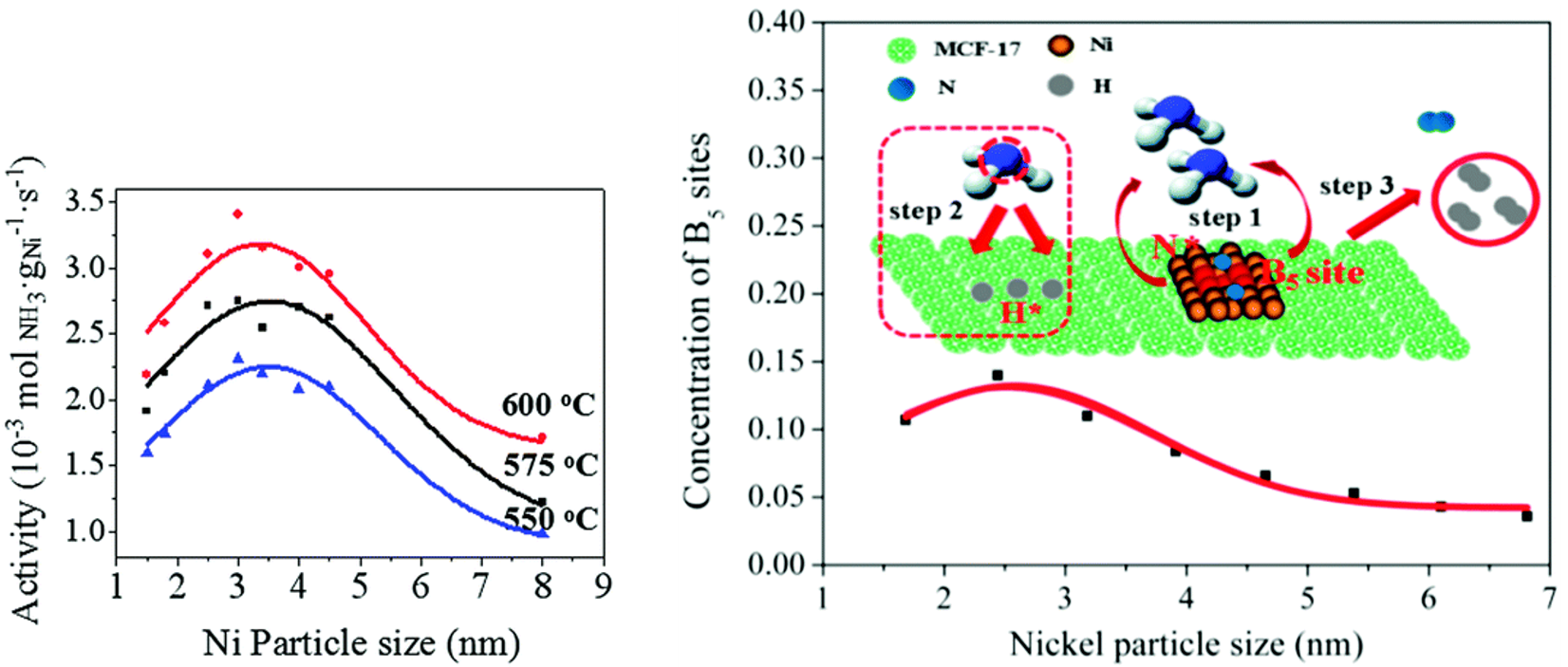
Figure 3. (a) NH3 decomposition activity of Ni/MCF-17, normalized to the Ni loading, for various reaction temperatures as a function of the average Ni NP size. Reaction conditions: P = 1 bar, pure NH3, GHSV = 6000 mL gcat−1 h−1. (b) The concentration of active B5 sites as a function of the average Ni NP size, with NH3 decomposition reaction mechanism on B5 sites of Ni catalysts. Step 1: NH3 chemisorption; step 2: consecutive dehydrogenation of NH3; step 3: associative desorption of H* and N* to form H2, respectively, N2. N* and H* denote adsorbed N and H atoms. Reproduced from Ref. [69] with permission from the Royal Society of Chemistry.
With regard to the application of colloidal synthesis for supported Co FTS catalysts, one of the first studies was performed by Melaet et al. [80], who prepared Co/TiO2 and Co/SiO2 with monodisperse Co NP sizes of ~10 nm. The activity towards FTS was twice as high for oxidized Co/TiO2 than for its reduced counterpart, while the contrary held true for Co/SiO2 (Figure 4a). In addition, this oxidation state influenced the product distribution, as more unsaturated products were formed for CoO/TiO2 (Figure 4b). In situ X-ray absorption spectroscopy (XAS), near edge X-ray absorption fine structure spectroscopy with total electron yield (NEXAFS-TEY), XRD and near-ambient pressure XPS (NAP-XPS) studies of Co/TiO2 indicated that this behavior originated from the reversible adsorbate-induced encapsulation of metallic Co by TiO2-x species, illustrated in Figure 4c. Upon H2 treatment at 450 °C, Co is fully reduced, yet encapsulated by TiO2, limiting the fraction of exposed active Co sites. Upon O2 treatment, this encapsulation is reversed, and an accessible, catalytically active “TiO2-wetted CoO phase” is formed. Exposure of this CoO material to H2 at 250 °C did not induce TiO2 encapsulation nor CoO reduction.
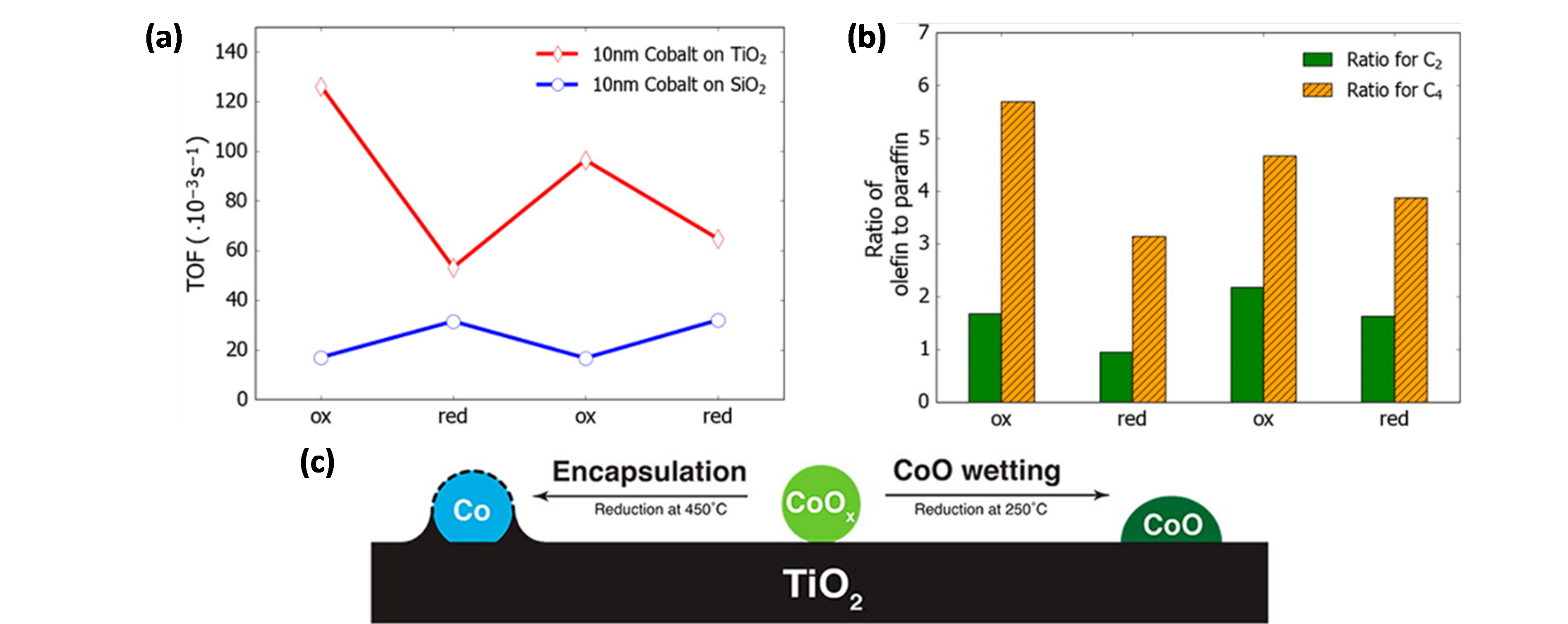
Figure 4. (a) TOF values for Co/TiO2 and Co/SiO2 FTS catalysts as a function of the oxidation state. (b) Olefin to paraffin ratios of C2 and C4 hydrocarbons for the Co/TiO2 FTS catalyst. (c) Schematic representation of the reversible TiO2 encapsulation of Co in Co/TiO2.“Ox” denotes oxidized Co, “red” reduced Co. Reduced Co was obtained via a H2 treatment. (H2: 10% (V/V), balance Ar) at 450 °C for 1 h, oxidized Co upon O2 treatment (O2: 20% (V/V), balance Ar) at 350 °C and maintained in a H2 atmosphere at 250 °C. Reaction conditions for (a) and (b): T = 250 °C, P = 5 atm, CO/H2 = 1/2, 24 h. Adapted with permission from Ref. [80]. Copyright 2014 American Chemical Society.
The colloidal synthesis of Co/TiO2 FTS catalysts was also reported by Delgado and co-workers [81] to investigate the influence of NP size (1.7–7 nm) on catalytic performance. Smaller sizes resulted in higher activity and catalyst stability, which were correlated respectively with the higher reducibility of the smaller NPs and a promoting effect of residual boron, stemming from the NaBH4 reducing agent used in the synthesis [82][83][84]. In other work [61][61], the same group concluded that, for a given NP size, the reducibility is affected by the polymer stabilizing agent used in the synthesis. of the six water-soluble polymers investigated, poly(2-ethyl-2-oxazoline) resulted in the highest FTS activity.
In the development of FTS catalysts with high selectivity towards olefins, i.e., FT to olefins (FTO), and their further conversion into aromatics, Krans et al. [85] prepared Na2S-promoted Fe NPs anchored onto H-ZSM-5, denoted as “FeP/Z”. For comparison, unpromoted Fe NPs were also deposited onto H-ZSM-5 (“Fe/Z”). Subsequent Na2S promotion of this Fe/Z yielded “Fe/Z-P”. Overall, the aforementioned materials exhibited low FTO activities due to Na2S overpromotion (Figure 5c). To avoid detrimental overpromotion effects, the aforementioned materials were washed with an ammonia nitrate solution for ion-exchange, yielding “FeP/Z-W” and “Fe/Z-P-W”. This washing step was also applied to Fe/Z, resulting in “Fe/Z-W”. Now, FeP/Z-W and Fe/Z-P-W outperformed unpromoted Fe/Z-W (Figure 5d), producing aromatics and having selectivities over 50%c towards C2–C4 fractions (Figure 5e). The methane selectivity was more than double for FeP/Z-W (40%c) than that for Fe/Z-P-W (15%c). Moreover, the promotion affected the particle growth during reaction. While notable Fe NP growth was observed for all promoted samples, whether or not washed, Fe NPs had grown most significantly for the FeP/Z catalysts (Figure 5a–b). This was ascribed to accelerated Ostwald ripening during FTO by the presence of promoters [86]. Consistently, inductively coupled plasma optic emission spectroscopy indicated the highest promoter-related Na and S content within FeP/Z, thus explaining the most notable NP growth in this sample. As expected from the lack of promoter elements in Fe/Z and Fe/Z-W, size distributions for these catalysts had changed insignificantly.
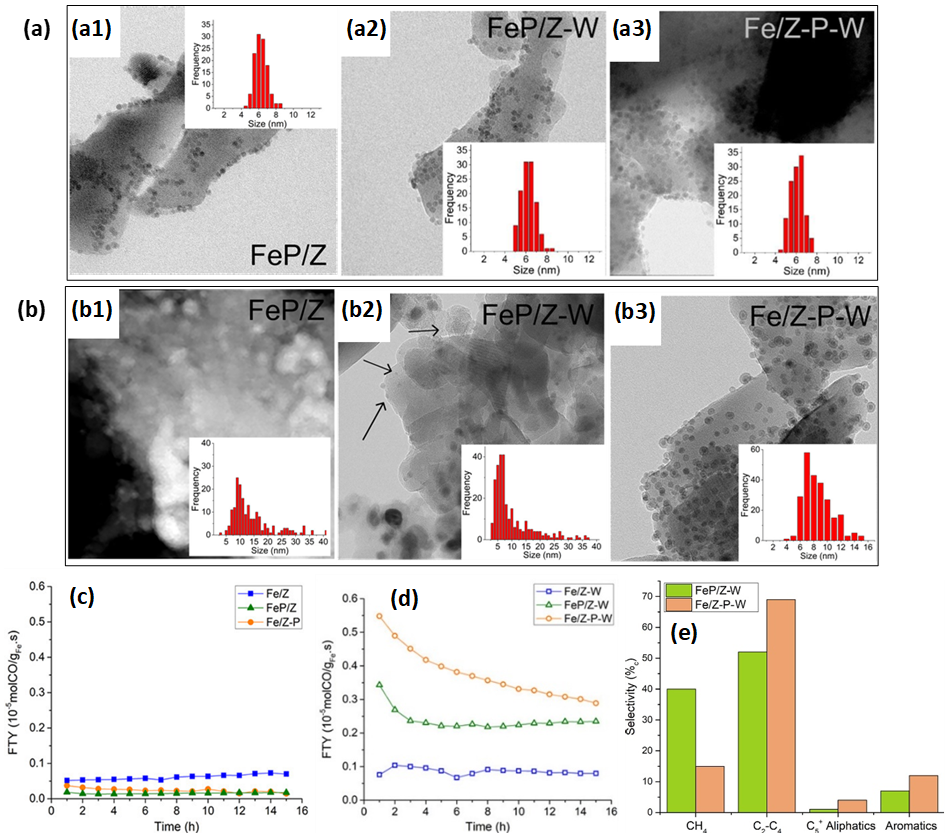
Figure 5. (a) TEM of as-prepared (a1) Fe/Z, (a2) FeP/Z, (a3) Fe/Z-P, (a4) Fe/Z-W, (a5) FeP/Z-W and (a6) Fe/Z-P-W. (b) Post-FTO TEM of (b1) Fe/Z, (b2) FeP/Z, (b3) Fe/Z-P, (b4) Fe/Z-W, (b5) FeP/Z-W and (b6) Fe/Z-P-W. Insets in (a) and (b) denote Fe NP size distributions. (c) Iron time yields (FTY, defined as moles of CO being converted per gram of iron per second) as a function of time-on-stream (TOS) for as-prepared, unwashed catalysts and (d) washed catalysts. Reaction conditions: T = 340 °C, P = 1 bar, H2/CO = 1/1 (V/V), CO conversion ~3–5%, GHSV = 5000 h−1. (e) Product selectivities of FeP/Z-W and Fe/Z-P-W in FTO conditions (T = 340 °C, P = 1 bar, H2/CO = 1 (V/V), GHSV: 4 200 h−1, TOS = 15 h). Adapted with permission from Ref. [85], https://pubs.acs.org/doi/abs/10.1021/acscatal.9b04380. Further permissions related to the material excerpted should be directed to the ACS.
CO hydrogenation into ‘short’ carbon chain products is another option to valorize syngas originating from alternative resources. In a study on tuning the shape of Rh/ZrO2 catalysts for CO hydrogenation, van Hoof et al. [87] examined the effect of capping agent removability. Only for oleylamine (OAm)-stabilized NPs could thermal reductive treatments remove the larger part (89%) of the capping agent. When trimethyl(tetradecyl)ammonium bromide (TTAB) or PVP were used, residual capping agent covered 38–68% of the active Rh sites, as calculated from TEM and H2 chemisorption experiments. However, the resulting decrease in activity relative to the OAm-capped samples was limited, being 20–27%. The authors ascribed this to the fact that TTAB and PVP blocked mostly “non-critical”, i.e., less active, sites or that the conditions used in the experiments to determine the fraction of sites blocked are not representative for those under real CO chemisorption conditions. Noteworthy is that this ‘capping agent blocking effect’ greatly overshadowed any observable shape-induced changes in the activity. It is therefore important that the capping agent and catalyst thermal treatment are chosen such that the agent is removed for unbiased structure-reactivity studies.
Other than CO hydrogenation, the hydrogenation of CO2 has been the topic of many CO2 utilization studies. In the selective hydrogenation of CO2 to methanol (MeOH), Lam et al. [88] reported the synthesis of “Cu/Zr@SiO2” catalysts, i.e., wherein isolated Zr(IV) sites are first grafted onto SiO2 (“Zr@SiO2”) and Cu NPs are subsequently dispersed onto this material, to facilitate investigation of the interfacial interactions between Zr surface sites and supported Cu NPs. This catalyst showed an enhanced MeOH formation rate and selectivity compared to analogously prepared Cu/ZrO2 and Cu/SiO2. In-situ XAS and ex-situ solid-state nuclear magnetic resonance (NMR) spectroscopy indicated this improvement arose from the promotional effect of Zr(IV) sites at the support periphery activating CO2 and hydrogenating reaction intermediates.
2.3. Bimetallic Supported Catalysts by Colloidal Synthesis
The use of a secondary element in NP synthesis offers multiple advantages over monometallic NPs. In a first instance, interactions between the elements can increase the catalytic activity, yield, stability, and resistance towards poisonous species [89][90][91]. In the case of precious metals, the use of secondary, inexpensive elements can reduce the overall catalyst cost. Furthermore, bimetallic NPs introduce composition (i.e., relative abundance of each element over the whole NP) and architecture (e.g., random alloy, core-shell) as additional degrees of freedom [92], which provides more opportunities for catalyst improvement. Given the tunability inherent to colloidal approaches in controlling NP parameters, and thereby their performance, research on bimetallic catalysts prepared via such techniques has received much attention.
2.3.1. Oxidation Reactions
In view of improving Au catalysts for CO oxidation by the addition of Cu, Destra et al. [93] studied AuCu catalysts (~10.2 ± 1.0 nm, in a fresh sample) on γ-Al2O3 and SiO2 supports, prepared via a colloidal co-reduction method. The catalysts’ performance was substantially influenced by oxidation (O2) and reduction (H2) cycles prior to reaction [94][95]. After each O2 cycle, the CO conversion in the subsequent activity test increased, up to 23% after three cycles (Figure 6a–b), and attaining 34% CO conversion after two subsequent reaction cycles without O2 pre-treatment (run #5 in Figure 6a–b). In contrast, after a single H2 cycle, the CO conversion already approached a maximum of 100% (Figure 6c,d). The significant enhancement after reduction by H2 was attributed to a more thorough purification of precursor-related poisonous Cl species under H2 than under O2. However, as reduction by H2 induced Au-Cu alloying and oxidation by O2 resulted in dealloying through the formation of separate CuOx phases, it was not excluded that the different performances after O2 and H2 cycles resulted from the formation of structures with quite different reactivities [96]. Notable herein is that, after the first oxidation and reduction cycle, no notable changes occurred in the material’s particle sizes relative to the fresh sample. The corresponding NP size distributions amounted to 10.2 ± 1.2 nm (fresh), 9.9 ± 1.2 nm (oxidized) and 10.1 ± 1.0 nm (reduced).
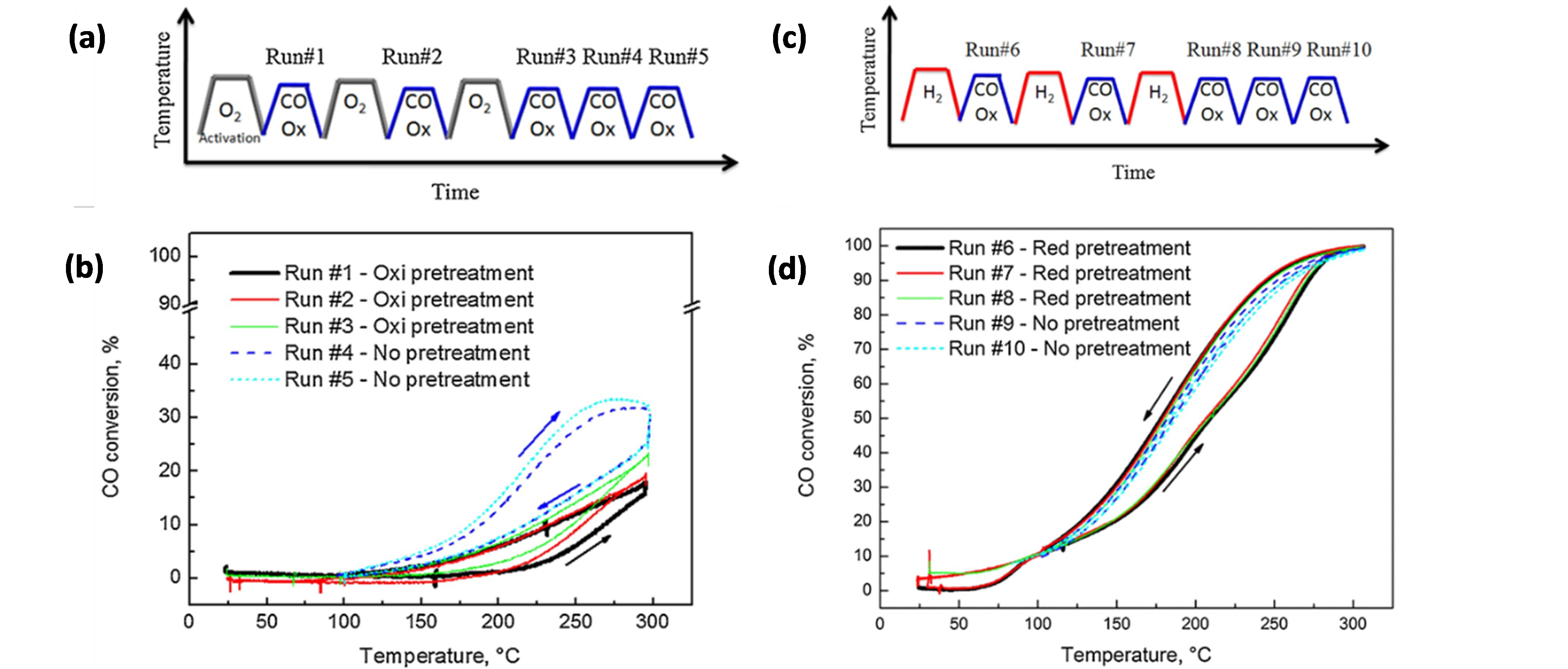
Figure 6. (a) Oxidation cycles coupled with CO oxidation tests applied to Au0.75Cu0.25/SiO2. (b) CO conversion results for the test protocol represented in (a). (c) Reduction cycles coupled with CO oxidation tests applied to Au0.75Cu0.25/SiO2. (d) CO conversion results for the test protocol represented in (c). Reaction conditions for (b) and (d): 1% CO (V/V), 6% O2 (V/V), He balance, GHSV = 3,000,000 Ncc/min (g of (Au + Cu)). Reprinted with permission from Ref. [94]. Copyright 2017 Elsevier.
Zaytsev et al. [97] prepared AuPd/γ-Al2O3 nanoalloys using various stabilizing agents (PVP, polyvinyl alcohol (PVA), Triton X-100 and AF-6 and AF-12 neonols) in the colloid synthesis. The CO oxidation performance of these catalysts was influenced most by the thermal treatment prior to reaction. For a given reaction temperature, calcination in dry air resulted in higher CO conversions than calcination in a H2 atmosphere or mere drying in air. This was ascribed to a more complete removal of the stabilizing agent, as corroborated by thermogravimetric analysis (TGA). However, calcination resulted in particle growth to 15–20 nm sizes, in contrast to the 3–6 nm nanocrystallite sizes of the catalyst prepared by drying. In addition, Triton X-100-stabilized NPs resulted in slightly higher activities, which was related to the stronger interaction of this surfactant with the support, thus enhancing NP immobilization and dispersion during reaction.
Nagy et al. [98] studied Ag as the secondary metal phase for SiO2-supported Au catalysts for their application in benzyl alcohol oxidation and CO oxidation. They assessed the influence of NP composition and catalyst pre-treatment, i.e., calcination and successive reduction, on the catalytic performance. For benzyl alcohol and CO oxidation, the activity as a function of the molar Ag/Au ratio reached a maximum for Ag/Au = 23/77 in both the calcined and reduced samples, thus indicating a synergistic effect compared to monometallic Ag or Au. Furthermore, the oxidation state of the metal had a significant influence on the activity. In benzyl alcohol oxidation, a successive thermal treatment resulted in an activity increase, while this had a more complex effect in the case of CO oxidation. These changes were attributed to the fact that the treatments resulted in the creation of different types and quantities of active sites. The exact underlying nature of these sites is to-date not fully clear, however.
In the class of total oxidation reactions, the catalytic combustion of alkanes forms an indispensable element in the context of pollution abatement and energy efficiency improvement. Due to its high energy density and potency as a greenhouse gas, methane is one of the designated reagents for combustion [99]. PtPd bimetallic catalysts are among the most important methane combustion catalysts. The combined use of Pt and Pd can lead to PtPd alloy formation, for which both positive [100][101] and negative [102][103] effects on combustion activity have been reported. Qu et al. [104] used the level of control granted by colloidal methods to devise a strategy to synthesize a PtPd/Mg-Al2O3 catalyst without PtPd alloy formation. When compared to a catalyst with the same metal loading prepared by WI, the colloidally prepared material already exhibited ~90% methane conversions at temperatures as low as 400 °C, while conversion values below 10% were obtained for the WI sample at the same temperature. The authors attributed this excellent performance to the formation of a Pt-PdO structure that enhances the redox properties of the catalyst and removes poisonous OH groups during combustion.
2.3.2. Hydrogenation Reactions
The addition of promoting elements is a known strategy for tuning the performance of FTS catalysts [105]. In an approach to study Co-Fe interactions and their effect on FTS performance, Ismail et al. [106] prepared monodisperse (5–7 nm) carbon nanotube (CNT)-supported CoFe catalysts via a nonhydrolytic colloidal method. The reduced catalyst displayed partial Janus-like alloy formation (Figure 7) and had higher activities and selectivities compared to monometallic counterparts. Moreover, through XRD and scanning TEM energy dispersive X-ray (STEM-EDX) analysis, the active phase in the bimetallic system for FTS was proposed to be a function of the employed reaction conditions, CoFe alloy and Fe- and Co-carbides being the identified phases in low- and high-temperature FTS, respectively.
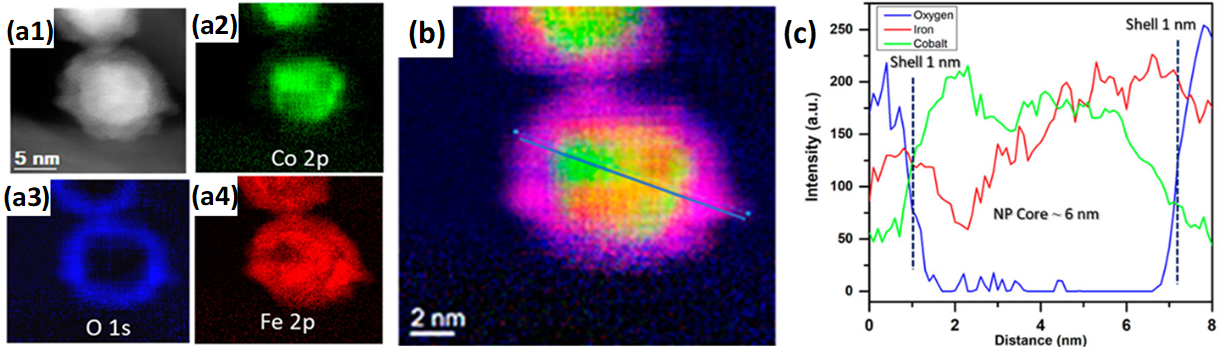
Figure 7. Janus-like CoFe alloy formation in CoFe/CNT FTS catalyst after reduction by H2. (a1) HAADF-STEM of CoFe/CNT catalyst; (a2)–(a4) corresponding EDX elemental mappings of (a2) Co, (a3) O and (a4) Fe. (b) Composite red-green-blue elemental mapping of Fe (red), Co (green) and O (blue), with EDX line scan pattern drawn on top. (c) EDX line scan profile corresponding to the pattern drawn in (b). Adapted with permission from Ref. [106], https://pubs.acs.org/doi/abs/10.1021/acscatal.8b04334. Further permissions related to the material excerpted should be directed to the ACS.
Dad et al. [107] studied the effect of Mn-promotion on Co via colloidally prepared CoMn supported on Stöber (SiO2) spheres. Apart from higher olefin selectivity, the catalyst proved more stable in FTS than unpromoted Co/SiO2. Not only the type of promoter but the NP architecture [108][109][110] can be used to tune the catalytic performance. Haghtalab et al. [111] applied a colloidal synthesis approach to prepare Co@Ru/γ-Al2O3 (supported core-shell NPs) catalysts with controlled Ru shell thickness. The electronic interaction between Co and Ru in the core-shell configuration resulted in higher CO conversion and selectivities towards desirable C5+ products than for monometallic Co and Ru catalysts. More so, a thicker Ru shell led to a higher increase in activity, illustrating the power of performance tunability achievable via colloidal techniques.
In the application of generating clean fuels [112][113]and chemical products [114] from biomass-derived resources, the direct production of dimethyl ether (DME) from syngas is an attractive option. Generally, such syngas-to-dimethyl (STD) catalysts consist of Cu/ZnO, i.e., the active phases in a standard methanol synthesis catalyst [115], and a dehydration component, e.g., γ-Al2O3 [116] or H-ZSM-5 [117]. To further the understanding of these catalysts through fundamental studies of well-defined materials, Gentzen and co-workers [118] reported a method starting from bimetallic CuZn colloidal NPs for the preparation of Cu/ZnO/γ-Al2O3 STD catalysts, which proved highly reproducible and resulted in uniform NP properties. Tunability of both the methanol formation and dehydration functionalities via this method, allowed achieving CO conversions up to 24% and DME selectivities up to 68%. The same synthesis protocol was used to systematically investigate the effect of the acidic properties of the dehydration component (γ-Al2O3, H-ZSM-5 or HY) on the performance in one-step conversion of syngas to DME and hydrocarbons [119]. For given reaction conditions, the CO conversion, selectivities towards DME and C1–C4 products, and the DME formation rate could be tuned by varying the ratio of Cu to acidic sites as well as the micropore area.
Based on the reported efficiency of PdZn phases for MeOH synthesis [120][121], Gentzen et al. [122] developed a colloidal protocol for the synthesis of Pd/ZnO-γ-Al2O3 as an alternative to the aforementioned CuZn-based STD catalyst. Most notably, STD activities, DME selectivity and catalyst stability were enhanced relative to conventional Cu/ZnO/γ-Al2O3. Following density functional theory (DFT) calculations and in situ and operando XAS, this was attributed to the presence of a stable intermetallic PdZn phase that is formed during catalyst activation and is the designated MeOH active component as part of the bifunctional STD catalyst.
Higher (C2+) alcohols are an equally viable CO hydrogenation product for a wide variety of day-to-day applications, e.g., fuels, cosmetics, polymers [123]. Bimetallic CuFe catalysts are a promising material for catalytic syngas conversion into such higher alcohols [124][125][126][127]. He et al. [128] created well-defined CuFe/CNT materials to study the structural factors that impact their performance in the formation of C2+ alcohols from syngas. They utilized pre-formed colloids prepared via stepwise- and co-reduction approaches and compared these with catalysts prepared through conventional WI. The catalyst prepared via co-reduction showed the highest C2+ alcohol selectivity (~21%). As the distance between Fe and Cu domains proved to be the smallest for this sample, as determined by high-angle annular dark-field scanning TEM (HAADF-STEM) imaging, it was proposed that the C2+ alcohol selectivity correlated with the proximity between the active Cu and Fe2C species. Quantum-chemical simulations corroborated this, as the energy barrier of the rate-determining step, determined as the C-C coupling to form CH3CHO (CH3 + HCO → CH3CHO), was higher when the contact between Fe and Cu species was less intimate.
For the purpose of investigating the promoting effect of Ru on Fe with respect to the selective hydrogenation of CO2 to hydrocarbons, Aitbekova et al. [129] synthesized well-defined Al2O3-supported “Ru/FeOx heterodimers” (Figure 8a) via the successive growth of iron oxide NPs (~13.1 nm) on pre-synthesized Ru NPs (~4.8 nm). Through in situ XAS studies, the authors proposed that the synergistic effect between Ru and Fe is implemented via hydrogen spillover from Ru to neighboring Fe. Furthermore, after a reductive activation pre-treatment, the heterodimers transformed into “Ru-Fe” core-shell structures. Post-reaction TEM, represented in Figure 8b, revealed this structure persists during the reaction. However, the formation of this relatively thick (~4.3 nm) Fe shell introduced a catalytic behavior similar to that of a monometallic Fe catalyst, thus virtually eliminating the effect of Ru. These considerations led to the design of an Al2O3-supported “Ru-FeOx” core-shell catalyst with a thinner shell (~1.2 nm) through an adapted stepwise reduction protocol. Yields for this catalyst were four times higher than for the heterodimer Ru-Fe variant (Figure 8c). Post-reaction TEM revealed no morphology changes in the catalyst with a thin Ru shell.
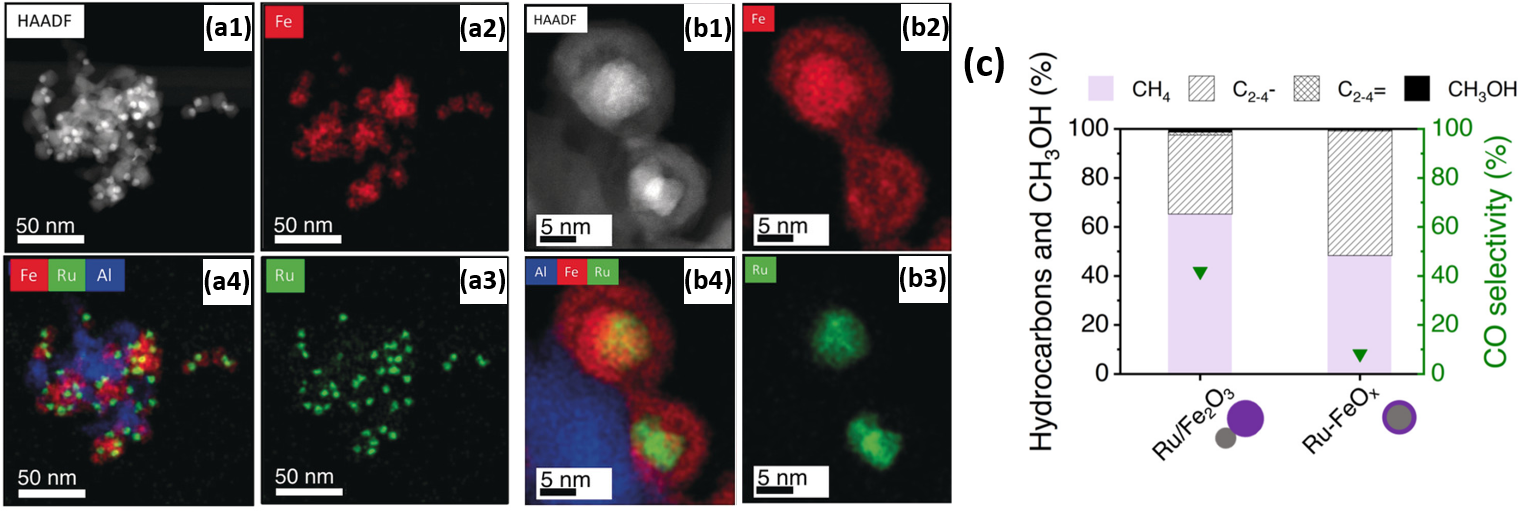
Figure 8. (a1) HAADF-STEM of Al2O3-supported Ru/Fe2O3 heterodimers after calcination; (a2)–(a4) corresponding EDX elemental mappings of (a2) Fe, (a3) Ru and (a4) composite Fe, Ru and Al mapping. (b1) HAADF-STEM of Al2O3-supported heterodimers after reduction by H2 at 300 °C and subsequent CO2 hydrogenation; (b2)–(b4) corresponding EDX elemental mappings of (b2) Fe, (b3) Ru and (b4) composite Fe, Ru and Al mapping. (c) Hydrocarbons and MeOH distributions and CO selectivities in CO2 hydrogenation for heterodimer Ru/Fe2O3/Al2O3 and core-shell “Ru-FeOx/Al2O3” catalysts reduced at 300 °C. Reaction conditions: 25% CO2 (V/V), balance H2, T = 300 °C, P = 6 bar, mcat = 30 mg, CO conversion = 18% (Ru/Fe2O3), 19% (Ru-FeOx). Reproduced with permission from Ref. [129]. Copyright 2019 John Wiley and Sons.
Within the selective hydrogenation reactions, the partial hydrogenation of alkynes via heterogeneous AuNi catalysts provides a cost-effective alternative to Pd-based materials [130]. Recently, Bruno and co-workers [131] reported a novel colloidal synthesis method for the preparation of Al2O3-supported AuNi NPs for application in 1-octyne partial hydrogenation. While bimetallic materials had activities and selectivities in between those of monometallic Ni and Au, their stability was significantly better than that of the constituent materials. The activity of Au and Ni catalysts halved within 20 h, whereas that of AuNi improved and remained stable for a week. Additionally, the tendency towards overhydrogenation was lower in the bimetallic catalysts, which was attributed to the presence of Au at the NP surface, suppressing H2 adsorption on Ni.
2.3.3. Dehydrogenation Reactions
The dehydrogenation of hydrocarbons to olefins is industrially exploited world-wide for the production of key building blocks for fuels, polymers, and fine chemicals [132]. With the goal of propane dehydrogenation (PDH), Sn-promoted Pt catalysts are particularly effective [133][134][135][136][137][138][139]. To investigate the extent of Sn promotion on Pt-based PDH catalysts, Kaylor and Davis [140] evaluated the influence of catalyst preparation method (colloidal versus IWI), Sn content and support (Al2O3 versus SiO2). As expected, colloidally prepared samples showed more monodisperse NP size distributions. The type of support affected the PDH performance similarly for both colloidally and IWI-prepared catalysts. Lower Sn loading resulted in less effective promoting effects in the case of Al2O3 than SiO2. When subjected to oxidative regeneration to remove coke formed during reaction, PtSn/Al2O3 recovered its activity while PtSn/SiO2 deactivated irreversibly due to significant PtSn dealloying by segregation of SnOx to the catalyst surface. These observations were ascribed to stronger MSI for Al2O3 than SiO2.
2.3.4. Reforming Reactions
Dry reforming of methane (DRM) involves the simultaneous conversion of the greenhouse gases methane and CO2 into syngas, which can serve as building block for a wide variety of reactions. Due to its environmental and industrial relevance, research effort has focused on the development of active, stable, and cost-effective DRM catalysts. In that regard, supported NiFe catalysts have proved effective in meeting these demands [141][142][143][144][145][146][148]. In an effort to improve the DRM activity by downsizing the NPs, Margossian et al. [149] applied a colloidal strategy in the preparation of uniform NiFe/Mg(Al)O catalysts with selected Ni- and Fe loadings. The initial activity of Ni/Mg(Al)O prepared by this route was 10 times higher than for a WI-prepared sample, but lost 89% of activity after 30 h time-on-stream (TOS). In contrast, monometallic Fe was inactive. While TOF values of an optimized Ni0.75Fe0.25 were slightly lower than for the monometallic Ni catalyst, it deactivated much more slowly, losing 31% of activity after 30 h TOS. For all bimetallic catalysts, higher reduction temperatures resulted in lower activities due to inactive Fe enriching the catalyst surface. At lower temperatures, NiFe alloy formation was more profound. Under DRM conditions, the constituent Ni0 is the active site for DRM, while neighboring Fe forms FeO, which allows for the decoking of Ni [141][146][147].
2.4. Gas-Phase Catalysis by Embedded NPs
While the use of colloids can indeed lead to high control over catalytic properties, deposited NPs obtained via this method typically suffer from structural instability upon exposure to thermal activation treatments and the reactive environment. For instance, metal sintering or dealloying can occur, compromising the structural integrity of as-synthesized colloids with correlated effects on the catalytic performance. In that regard, “embedding” the colloidal NPs within a support proves an interesting solution. By encasing the particles, their mobility is restricted, such that sintering-induced deactivation is mitigated [150]. The support’s porosity, created by thermal and/or chemical treatments [151][152][153], ensures the mass transfer of reagents and products. More so, tailoring the porosity of this supporting phase allows adapting the accessibility and rate at which certain molecules are transported, thus affecting the product selectivity and reaction rate [25].
Different embedment architectures can be distinguished based on the spatial distribution of the active phase and the support. Common ones are represented in Figure 9. In core-shell architectures [24][25], the core consists of the active phase which is completely encapsulated by a porous oxide shell. Yolk-shell is another established option [24][154]. Analogous to a bird’s egg wherein an egg yolk is surrounded by albumen and a hard shell, each individual NP in this architecture is successively surrounded by a cavity and a porous shell. As the NP core is enveloped by a relatively homogeneous environment, this makes the NP’s active sites more accessible for reaction than in core-shell architectures. Furthermore, the use of a core with multiple shells is possible, as are many other variations [155]—a full enumeration of which is beyond the scope of this review. Apart from the deposition of porous layers onto each NP, colloidal NPs can also be embedded—partially or completely—within a support matrix [21]. The latter is thus in contrast with yolk/core-shell structures, as this does not lead to the individual encapsulation of NPs with a separate support layer, but rather to multiple NPs encapsulated in a common support phase. For a detailed description of the synthesis of these different architectures, the reader is referred to dedicated literature [21][24][25][27][155][156].

Figure 9. Common “embedded” NP catalyst architectures. Purple denotes the active phase, grey the support phase.
The wide array of embedment techniques for colloidal NPs does not only offer a means of stabilizing NPs, but adds additional tunability to the design of solid catalysts by adjustment of the support’s architecture and porosity. In this section, recent gas-phase catalysis applications of support-embedded catalysts prepared via a colloidal template are discussed.
2.4.1. Oxidation Reactions
As stated before, CO oxidation is a probe reaction used to investigate the effect of composition and structure of the active phase under reaction conditions. In the case of embedded nanomaterials, it is therefore ideal to investigate the degree of structural stability induced by the encapsulation phase. Due to the structure-insensitivity of CO oxidation over Pd catalysts, Seo et al. [157] specifically opted for this reaction to evaluate the effect of aging Pd@SiO2 core-shell materials in air. Most notably, a better oxidation performance was observed when the material was aged at 800 °C instead of at 500 °C (Figure 10a). This originated from the partial redispersion of the ~4 nm Pd cores into smaller ~2 nm NPs within the SiO2 shells, which only occurred at temperatures above 800 °C, as revealed by in situ TEM imaging (Figure 10b–d). At 500 °C, the Pd mobility was too low to allow for this redispersion, such that the NP retained its original morphology. In contrast, a supported Pd/SiO2 catalyst underwent notable sintering at these temperatures.

Figure 10. (a) CO oxidation light-off curves for Pd@SiO2 after aging in air at 500 °C (blue) and at 800 °C (red). Reaction conditions: 2 °C/min, 1% (V/V) CO, 1.5% (V/V) O2, balance N2, total flow rate = 200 mL min−1, mcat = 60 mg. (b)–(d) In situ (S)TEM of Pd@SiO2 after aging in 150 Torr O2 at (b) 200 °C for 1h, (c) 500 °C for 30 min and (d) 800 °C for 30 min. Reprinted with permission from Ref. [157]. Copyright 2018 Elsevier.
For the purpose of embedding NPs within a support material, catalyst synthesis via a raspberry colloid-template (RCT) is a promising method [158][159]. Such RCT materials are prepared by binding metallic NPs onto the surface of colloidal polymeric particles, which creates colloids with a “raspberry” morphology. Following the spontaneous assembly of these raspberry colloids, the interstitial spaces of this assembly are filled with metal oxide matrix precursor material. By subsequently subjecting this system to a heat treatment, the polymeric template is removed, leaving only a porous metal oxide with continuous, interconnected pores, decorated with metal NPs. Luneau and co-workers [160] studied PdAu NPs with dilute, i.e., low, Pd concentration (maximum 0.09 at%), partially embedded in raspberry colloid-templated-SiO2 (RCT-SiO2). They demonstrated the material’s stability under CO oxidation (Figure 11a). This latter aspect was corroborated by in situ XAS measurements as no notable changes in either Pd K or Au L3 edge features were observed under reactive conditions, indicating no net change occurred to the initial PdAu alloy state (Figure 11b–d).
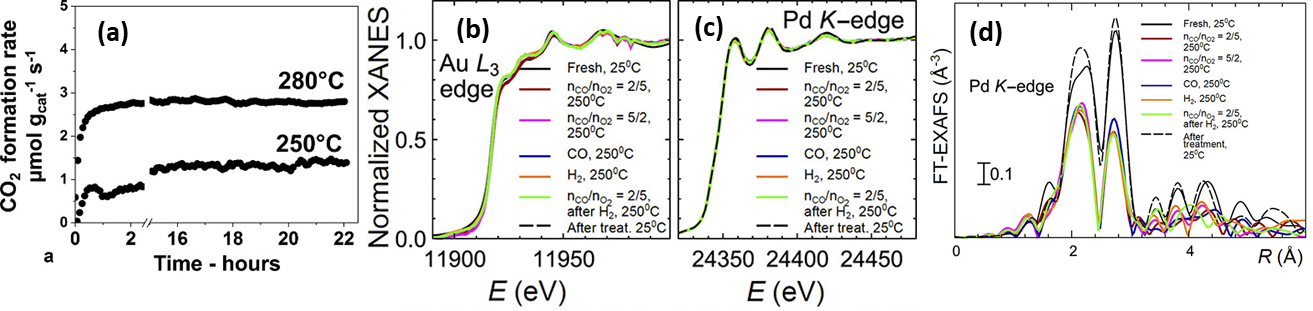
Figure 11. (a) CO oxidation stability test results of Pd0.09Au0.91 RCT-SiO2. Reaction conditions: 5% (V/V) CO, 10% (V/V) O2, He balance; GHSV = 2000 h−1, total flow rate = 25 mL min−1 ; mcat = 40 mg. (b) In situ XANES spectra for the Au L3 edge and (c) the Pd K edge for Pd0.09Au0.91 RCT-SiO2 under reaction conditions. (d) Corresponding Fourier-transformed k2-weighted EXAFS spectra for the Pd K edge. Adapted with permission from Ref. [160]. Copyright 2019 American Chemical Society.
Kim et al. [161] evaluated Pt@SiO2 core-shell materials in both CO and CH4 oxidation as a means to investigate its stability in low (<300 °C) and high (>500 °C) temperature regimes, respectively. While Pt@SiO2 achieved lower CO oxidation activity compared to supported Pt/SiO2, which was attributed to a lower number of exposed sites in the embedded material, similar activation energies were obtained for both materials, indicating an identical oxidation mechanism. Under CH4 oxidation conditions, Pt/SiO2 agglomerated, while Pt@SiO2 largely preserved its morphology, resulting in higher activity and stability. Still, deactivation occurred for Pt@SiO2, albeit to a lesser extent than in Pt/SiO2. This was hypothesized to be caused by temperature-induced shell degradation and Pt NP agglomeration.
Given the high operating temperatures of catalytic combustion reactions, embedding NPs within oxide materials is an attractive way to enhance catalyst stability for these reactions. Habibi et al. [162] tested SiO2-encapsulated colloidal PtPd alloy NPs in wet (5 mol% H2O vapor) and dry catalytic CH4 lean combustion. The catalyst performance remained stable after 70 h under combustion conditions. Additionally, the catalysts were tested for stability after hydrothermal aging, which comprised a 50 h thermal treatment using the wet combustion feed. Hydrothermally aged SiO2-encapsulated structures achieved two- and ten-times higher conversions with respect to similarly treated PdPt/Al2O3 and PdPt/Al2O3 prepared via IWI. The enhanced performance was correlated with the higher dispersion of PtPd NPs within the SiO2 shell after aging. For the purpose of enhancing the thermal stability of Pt toluene combustion catalysts without inducing severe mass transport limitations, Pei and co-workers [163] developed an in situ synthesis method to partially embed Pt NPs into three-dimensionally ordered macroporous (3DOM) Mn2O3, whereby the Mn2O3 structure consists of a three-dimensional structure with ordered spherical macropores (>50 nm) which is prepared by a colloidal template method [164]. 2.3 wt% Pt/Mn2O3-3DOM exhibited the best catalytic performance. Due to the confining nature of this embedment, the stability, as tested for 60 h TOS, proved to be better than that of a similar material prepared via colloid adsorption. Consistent with these results, the average NP size of the embedded Pt catalyst changed insignificantly from 4.3 nm to 4.7 nm.
Recently, Shirman et al. [159] made progress in catalyst design for selective alcohol oxidation by applying a partial embedment of AgAu NPs in RCT-SiO2 and assessing their performance in the oxidative coupling of MeOH and EtOH to produce esters. The robustness of these catalysts was proved by their stable catalytic performance. In accordance, TEM imaging confirmed the absence of NP sintering and agglomeration both after calcination and repeated activity tests with 40 h TOS.
2.4.2. Hydrogenation Reactions
While SiO2 encapsulation is an option to prevent the detrimental effects of NP agglomeration and sintering in Fe FTS catalysts, SiO2 suffers from low hydrothermal stability and can have adverse effects on the catalytic properties upon interaction with Fe [165][166]. In that regard, modification of SiO2 through the addition of promoters presents an opportunity. To weaken the Fe-SiO2 interactions, Ni et al. [167] applied graphitic carbon-promoted SiO2 (SiO2-GC). Core-shell Fe@SiO2-GC catalysts with optimized composition could achieve higher CO conversions and C2–C4 selectivities than an Fe@SiO2 core-shell catalyst after 100 h TOS. Conversion, stability, and selectivity improvements were attributed to the GC-promoted SiO2 shell. Due to the enhanced hydrothermal robustness by GC-promotion, the shell integrity was conserved under FTO conditions, such that Fe confinement and high Fe dispersion were maintained even over longer TOS. Moreover, the narrow pore size distribution of the SiO2-GC shell exerted a spatial-restricting effect that inhibited the formation of longer carbon chains and favored the formation of shorter (C2–C4) chain lengths.
In the selective hydrogenation of CO into higher alcohols via CuFe catalysts, embedding NPs in porous metal oxides is ideal to maintain intimate contact between Cu and Fe species during reaction. A facile method for the encapsulation of pre-synthesized CuFe NPs in SiO2 shells was developed by Huang et al. [168]. The prepared structures were spherical in nature (average diameter 80–90 nm) and consisted of multiple CuFe cores in a shared SiO2 shell. In CO hydrogenation activity studies, CuFe@SiO2 materials outperformed unsupported CuFe NPs in terms of alcohol selectivity and CO conversion after 252 h TOS. Comparative pre-use and post-mortem TEM studies of the embedded material revealed the formation of dual functional Cu–χ-Fe5C2 during reaction, a phase which was maintained, i.e., without sintering or Cu-Fe phase separation, due to the restricting nature of the shell, thus explaining its enhanced performance.
Ilsemann et al. [169] reported Co@SiO2 and Co@silicalite-1 catalysts for the methanation of CO, CO2 and CO/CO2 mixtures. Compared to Co/SiO2 prepared by WI, the colloidally-prepared embedded materials exhibited increased CH4 formation rate and selectivity in the order Co/SiO2 < Co@silicalite-1 < Co@SiO2. These observations were linked to the confinement by the silica phase, which is most pronounced in Co@SiO2. It was proposed that this effect stimulates the adsorption and hydrogenation of CO intermediates in the reactive environment near the active Co phase. Carbon-related deactivation occurred for all catalysts under methanation conditions. No sintering was observed for the core-shell catalysts. This is in contrast with Co/SiO2, whereby sintering induced an increase of the average NP size, as pre-and post-reaction NP sizes amounted to 39 nm and 44 nm, respectively. Hence, the absence of sintering in Co@SiO2 justifies the use of encapsulation for stability enhancement.
Analogous to their previously mentioned application in CO oxidation [160], Luneau et al. [170] demonstrated the use of Pd-dilute PdAu RCT-SiO2-embedded nanocatalysts for the selective hydrogenation of 1-hexyn into 1-hexene. A Pd0.04Au0.96 catalyst, i.e., with optimized composition, proved superior to monometallic Pd as it achieved high alkene selectivities (>90%) at high alkyne conversions (~80%) for an extended reaction time of 30 h (Figure 12a). The authors attributed the selectivity results to the dilute surface concentration of Pd active sites, which would favor partial hydrogenation [171][172][173][174]. Comparative (S)TEM(-EDX) analysis, represented in Figure 12b–d, revealed neither sintering nor Pd-Au phase separation after calcination or reaction, thus demonstrating the stabilization effect of the catalyst’s architecture. While carbon formation was proved by temperature-programmed oxidation, no detrimental effects on catalytic performance were observed.
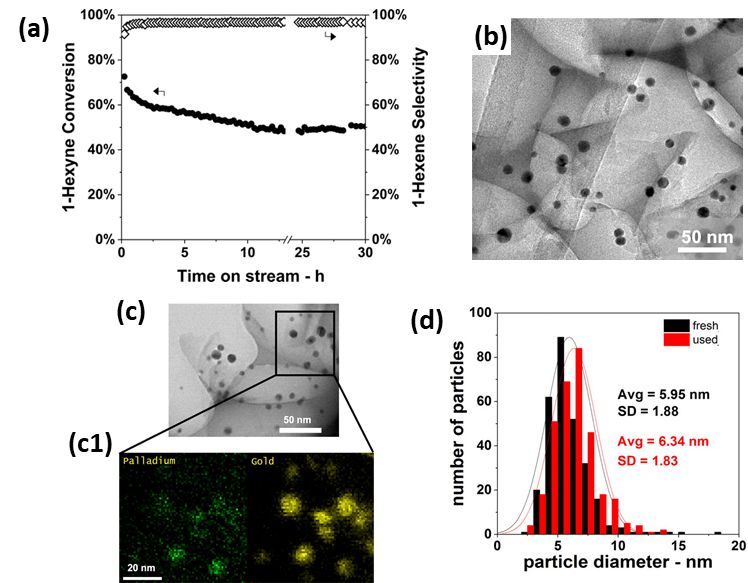
Figure 12. (a) 1-hexyne conversion and 1-hexene selectivities in 1-hexyne partial hydrogenation as a function of TOS for Pd0.04Au0.96 RCT-SiO2. Reaction conditions: T = 90 °C, P = 1 atm, 1% (V/V) 1-hexyne; 20% (V/V) H2, He balance, mcat = 20 mg, total flow rate = 50 mL min−1, GHSV = 3800 h−1. (b) TEM of fresh Pd0.04Au0.96 RCT-SiO2. (c) TEM of Pd0.04Au0.96 RCT-SiO2 after the stability test in (a); (c1) STEM-EDX color mapping of (c); green: Pd; yellow: Au. (d) Particle size distributions of fresh and used Pd0.04Au0.96 RCT-SiO2. Adapted with permission from Ref. [170]. Copyright 2020 American Chemical Society.
Stability improvement by embedding NPs is a promising tactic to further advance the industrial and environmental potential of catalysts for CO2 hydrogenation towards MeOH. A first successful synthesis of core-shell Cu@mesoporous SiO2 (m-SiO2) and Cu/ZnO@m-SiO2 catalysts was reported by Yang et al. [175]. Both Cu and Cu/ZnO NP centers conserved their monodisperse (~5 nm) size distribution after calcination, reduction, and reaction, as opposed to a reference Cu/m-SiO2 sample, where notable sintering occurred. As a result of the high metal dispersion in the core-shell architectures and their anti-sintering properties, these excelled in terms of conversion, MeOH selectivity and stability. Additionally, Cu/ZnO@m-SiO2 materials achieved twice the selectivities of monometallic Cu@m-SiO2 due to the introduction of additional basic sites by ZnO.
As an alternative to the aforementioned Cu/ZnO materials, Shi et al. [176] synthesized CuIn@SiO2 core-shell materials with superior anti-segregation properties. In activity tests, the catalytic performance was superior to that of CuIn/SiO2, both initially and after 100 h TOS. These results were attributed to both the isolating nature of the shell as well as to the Cu-In interactions within the core promoting Cu dispersion, metal reducibility, alloy formation, and oxygen vacancy formation, and thereby CO2 activation and hydrogenation.
2.4.3. Reforming Reactions
As hydrocarbon reforming is performed at high temperatures, one of the intrinsic challenges of these reactions is the rapid catalyst deactivation due to coke formation and sintering of the active phases. To counter these degradation effects, the design of embedded reforming catalysts has been studied extensively over the past decade. In view of the in-depth review on this topic written by Li et al. [150] at the end of 2018, only more recently published work will be covered in this section.
Among monometallic non-noble DRM catalysts, Ni materials are by far the most interesting as they exhibit higher activities compared to Co- or Fe-based DRM catalysts. In result, encapsulated Ni materials have been the subject of many studies [177]. Continuing from their previously reported colloidal protocol for the preparation of sinter-free and carbon-resistant core-shell-structured Ni@SiO2 [178], Zhang and co-workers [179] evaluated the effect of Ni NP size and MSI on the catalyst’s performance in DRM. To this end, catalysts with average Ni NP sizes of 1.4, 1.9 and 2.6 nm, as determined via TEM after activation, were prepared by employing calcination temperatures of 500, 600 and 700 °C, respectively. Reduction temperature analysis via H2-TPR indicated the strongest MSI occurred for higher calcination temperatures. The catalytic performance of the sample with intermediate NP size and intermediate MSI strength was superior in overall reforming activity and stability indicating that the DRM performance in these catalysts is both function of NP size and MSI. The group also demonstrated the stability of the same Ni@SiO2 catalyst in methane steam reforming (SRM) after 50 h TOS [180]. Additional TEM and XRD characterization revealed that the architecture conserved the initial morphology after SRM up to 750 °C. Similar structural and catalytic stabilities were observed when the catalyst was subjected to DRM at identical temperatures. However, the spent catalyst contained a higher carbon content after DRM than after SRM, which was attributed to the stronger carbon gasification power of H2O relative to CO2.
2.4.4. Water-Gas Shift
As WGS is one of the prime reactions for H2 production [181], improved design of WGS catalysts is of high importance for industry. While Ni [182] is a cost-effective alternative to noble metal-based [183] catalysts, it suffers from two intrinsic drawbacks under WGS conditions [184]: simultaneous CH4 formation, which restricts the H2 yield; and sintering of Ni. As a solution to these problems, Gao et al. [185] synthesized core-shell hexagonal boron nitride (h-BN)-encapsulated Ni NPs through a facile thermal NH3 treatment of PVP-capped Ni NPs. These core-shell Ni@h-BN catalysts retained their particle size under thermal treatments up to 850 °C and showed high WGS activity at low temperatures (~250 °C). In a stability test, the catalyst’s CO conversion remained stable at 90–92% during 30 h TOS, indicating no deactivation by sintering or coke formation. In that time, very little CH4 formation was observed, proving the beneficial role of the h-BN shell in increasing the H2 yield. Based on activity data and H2O pulse chemisorption experiments, it was suggested that h-BN mitigates methanation by inducing H2O dissociation at the h-BN surface or at the core–shell interface.
3. Conclusions and Outlook: Colloidal Synthesis Applied to Heterogeneous Catalysis
The creation of well-defined NPs supported on porous supports is fundamental for the rational design of catalysts. In that regard, bottom-up colloidal synthesis techniques have been successful in creating such uniform materials in view of their application in the gas-phase catalysis of various reaction families. As demonstrated in this review, studies have focused on supported monometallic, bimetallic NPs, as well as particulates embedded into the support or coated with porous oxide overlayers. Herein, colloidal synthesis has proven its worth in fundamental studies whereby the role of structure, composition, and the nature of the active site have been elucidated.
The controlled synthesis of supported NPs and their catalytic applications is an ever-growing discipline, such that future improvements are expected regarding the synthesis of novel materials, their characterization and the technologies involved in their fabrication.
Not taking the support or possible oxidic overlayers into account, controlled materials for gas-phase catalysis are to-date limited to mono- and bimetallic materials. While some tri- and multimetallic supported materials have been prepared using colloids [35], associated protocols are not as abundant as for mono- and bimetallic materials. In addition, most studies are focused on the synthesis of these materials rather than on applying them. In the select application studies that exist, gas-phase catalysis has not been researched [35][186][187][188]. Hence, future studies may consist in (1) the synthesis of novel tri- and multimetallic supported catalysts via colloidal synthesis and (2) their catalytic applications. Although various mono- and bimetallic systems have been explored through the use of controlled synthesis techniques, there are still many industrially relevant mono- and bimetallic catalysts which have only been examined via conventional and uncontrolled synthesis techniques. As a result, these latter catalysts can profit from the use of colloidal synthesis in extending the fundamental understanding of their properties and aid in the rational design of desirable catalytic sites. Of course, this necessitates the further development of colloidal protocols that can effectively create the above-mentioned materials. With the goal of cost minimization, related future work is best focused on non-noble metals.
In the fabrication of the abovementioned materials, general directions can be given for advanced synthesis strategies, that allow improved control over the nanoparticulate materials. One such strategy includes the exploitation of MSI. It has been demonstrated in this review that MSI play a pivotal role in determining the catalytic properties both during synthesis and reaction, e.g., by mitigating NP migration through strong interaction between support and metal. Nevertheless, systematic knowledge of the factors influencing MSI is lacking to-date, which instigates dedicated studies to investigate these factors in order to allow tailoring MSI for improved control of the catalytic performance. The use of embedded or coated NPs presents another opportunity. While such materials have proved effective in improving catalytic stability relative to bare NPs, the coating layers often limit access to the surface of the metals contained within. Hence, future work should focus on overcoming this shortcoming, for example, through the development of colloidal synthesis protocols which do not introduce this bottleneck. This requires the development of synthesis methods to further enhance the control over the spatial distribution, architecture and porosity of the oxide phase surrounding the active NPs.
As illustrated in this review, the use of established ex situ, in situ and operando techniques in catalytic studies with controlled NPs has already proved its value in attaining unrivaled fundamental understanding of the factors that influence catalytic performance. To bring this understanding via these fine-tuned materials to the next level, studies must move more towards in situ and operando techniques to allow unbiased tracking of geometrical and electronic changes that take place during the reaction and correlate these with the catalytic performance. However, as the active sites are only present in small quantities and their signature is often dominated by larger spectator contributions, it remains a challenge for in situ and operando characterization to investigate the real catalyst species with high selectivity. In that regard, the application of stimulated changes in the reactive atmosphere via modulation-excitation (ME) [189] has received strong attention to selectively identify the active sites on supported NPs. While ME has been successfully applied to XAS [190][191], as well as infrared spectroscopy [192], Raman spectroscopy [193] and XRD [194], it has to-date been little employed to study controlled materials prepared by colloidal synthesis. Hence, there is much untapped potential for well-defined systems through such in situ/operando experiments. Moreover, in situ kinetic studies that are (partially) transient in nature, such as temporal analysis of products (TAP) [195] or steady-state isotope transient kinetic analysis (SSITKA) [196], are little to not reported for well-defined materials [197]. Nevertheless, applying these techniques to controlled materials can give additional fundamental catalytic insights, especially when combined with the above-mentioned characterization techniques.
While catalysts fabricated via colloidal approaches are promising, related synthesis procedures are currently limited to lab-scale batch processes. Large-scale colloidal synthesis is hampered by the high cost of commonly used organometallic precursors, the large solvent volumes used to obtain high particle dilution and the lack of scalable NP stabilization methods [21]. Hence, studies focusing on upscaling these approaches are an interesting point for future research.
Even if commercial scale-up of controlled catalyst synthesis is never attained for colloidal synthesis, the method has, as demonstrated in this review, already proved its worth in research for a wide variety of fundamental studies. It is thus expected that this method shall continue to be employed for future studies, not only for academic purposes, but also to examine improvements for potential future industrial applications.
References
- Armor, J.N. A history of industrial catalysis. Catal. Today 2011, 163, 3-9, doi:https://doi.org/10.1016/j.cattod.2009.11.019.
- White, R.J.; Luque, R.; Budarin, V.L.; Clark, J.H.; Macquarrie, D.J. Supported metal nanoparticles on porous materials. Methods and applications. Chem. Soc. Rev. 2009, 38, 481-494, doi:10.1039/B802654H.
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687-6718, doi:10.1021/cr500486u.
- An, K.; Somorjai, G.A. Nanocatalysis I: Synthesis of Metal and Bimetallic Nanoparticles and Porous Oxides and Their Catalytic Reaction Studies. Catal. Lett. 2015, 145, 233-248, doi:10.1007/s10562-014-1399-x.
- Pan, C.-J.; Tsai, M.-C.; Su, W.-N.; Rick, J.; Akalework, N.G.; Agegnehu, A.K.; Cheng, S.-Y.; Hwang, B.-J. Tuning/exploiting Strong Metal-Support Interaction (SMSI) in Heterogeneous Catalysis. J .Taiwan Inst. Chem. Eng. 2017, 74, 154-186, doi:https://doi.org/10.1016/j.jtice.2017.02.012.
- Penner, S.; Armbrüster, M. Formation of intermetallic compounds by reactive metal–support interaction: a frequently encountered phenomenon in catalysis. ChemCatChem 2015, 7, 374-392.
- Cargnello, M.; Fornasiero, P.; Gorte, R.J. Opportunities for Tailoring Catalytic Properties Through Metal-Support Interactions. Catal. Lett. 2012, 142, 1043-1048, doi:10.1007/s10562-012-0883-4.
- Cao, S.; Tao, F.; Tang, Y.; Li, Y.; Yu, J. Size- and shape-dependent catalytic performances of oxidation and reduction reactions on nanocatalysts. Chem. Soc. Rev. 2016, 45, 4747-4765, doi:10.1039/C6CS00094K.
- Van Santen, R.A. Complementary structure sensitive and insensitive catalytic relationships. Acc. Chem. Res. 2009, 42, 57-66.
- Den Breejen, J.; Radstake, P.; Bezemer, G.; Bitter, J.; Frøseth, V.; Holmen, A.; de Jong, K.d. On the origin of the cobalt particle size effects in Fischer− Tropsch catalysis. J. Am. Chem. Soc. 2009, 131, 7197-7203.
- Somorjai, G.A.; Park, J.Y. Colloid Science of Metal Nanoparticle Catalysts in 2D and 3D Structures. Challenges of Nucleation, Growth, Composition, Particle Shape, Size Control and Their Influence on Activity and Selectivity. Top. Catal. 2008, 49, 126-135, doi:10.1007/s11244-008-9077-0.
- Somorjai, G.A.; Park, J.Y. Molecular Factors of Catalytic Selectivity. Angew. Chem. Int. Ed. 2008, 47, 9212-9228, doi:10.1002/anie.200803181.
- An, K.; Somorjai, G.A. Size and shape control of metal nanoparticles for reaction selectivity in catalysis. ChemCatChem 2012, 4, 1512-1524.
- Goodman, D.W.; Kelley, R.D.; Madey, T.E.; Yates, J.T. Kinetics of the hydrogenation of CO over a single crystal nickel catalyst. J. Catal. 1980, 63, 226-234, doi:https://doi.org/10.1016/0021-9517(80)90075-5.
- Oh, S.H.; Fisher, G.B.; Carpenter, J.E.; Goodman, D.W. Comparative kinetic studies of COO2 and CONO reactions over single crystal and supported rhodium catalysts. J. Catal. 1986, 100, 360-376, doi:https://doi.org/10.1016/0021-9517(86)90103-X.
- Oosterbeek, H. Bridging the pressure and material gap in heterogeneous catalysis: cobalt Fischer–Tropsch catalysts from surface science to industrial application. Phys. Chem. Chem. Phys. 2007, 9, 3570-3576, doi:10.1039/B703003G.
- Bridging the pressure and material gap in heterogeneous catalysis. Phys. Chem. Chem. Phys. 2007, 9, 3459-3459, doi:10.1039/B706675A.
- Marceau, E.; Carrier, X.; Che, M.; Clause, O.; Marcilly, C. Ion exchange and impregnation. In Handbook of Heterogeneous Catalysis, Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds. Wiley-VCH Verlag: Weinheim, Germany, 2008; Vol. 1, pp. 467-484.
- Behrens, M. Coprecipitation: An excellent tool for the synthesis of supported metal catalysts – From the understanding of the well known recipes to new materials. Catal. Today 2015, 246, 46-54, doi:https://doi.org/10.1016/j.cattod.2014.07.050.
- Geus, J.W.; van Dillen, A.J. Preparation of supported catalysts by deposition–precipitation. In Hand. Heter. Cat.: Online, Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds. Wiley-VCH Verlag: Weinheim, Germany, 2008; Vol. 1, pp. 428-467.
- Jia, C.-J.; Schüth, F. Colloidal metal nanoparticles as a component of designed catalyst. Phys. Chem. Chem. Phys. 2011, 13, 2457-2487, doi:10.1039/C0CP02680H.
- Tao, A.R.; Habas, S.; Yang, P. Shape Control of Colloidal Metal Nanocrystals. Small 2008, 4, 310-325, doi:10.1002/smll.200701295.
- Collins, G.; Holmes, J.D. Engineering Metallic Nanoparticles for Enhancing and Probing Catalytic Reactions. Adv. Mater. 2016, 28, 5689-5695, doi:10.1002/adma.201503970.
- Li, G.; Tang, Z. Noble metal nanoparticle@metal oxide core/yolk–shell nanostructures as catalysts: recent progress and perspective. Nanoscale 2014, 6, 3995-4011, doi:10.1039/C3NR06787D.
- Zhang, Q.; Lee, I.; Joo, J.B.; Zaera, F.; Yin, Y. Core–Shell Nanostructured Catalysts. Acc. Chem. Res. 2013, 46, 1816-1824, doi:10.1021/ar300230s.
- Lee, I.; Albiter, M.A.; Zhang, Q.; Ge, J.; Yin, Y.; Zaera, F. New nanostructured heterogeneous catalysts with increased selectivity and stability. Phys. Chem. Chem. Phys. 2011, 13, 2449-2456, doi:10.1039/C0CP01688H.
- Tian, H.; Li, X.; Zeng, L.; Gong, J. Recent Advances on the Design of Group VIII Base-Metal Catalysts with Encapsulated Structures. ACS Catal. 2015, 5, 4959-4977, doi:10.1021/acscatal.5b01221.
- Bönnemann, H.; Richards, R.M. Nanoscopic metal particles− synthetic methods and potential applications. Eur. J. Inorg. Chem. 2001, 2001, 2455-2480.
- Lu, Z.; Yin, Y. Colloidal nanoparticle clusters: functional materials by design. Chem. Soc. Rev. 2012, 41, 6874-6887, doi:10.1039/C2CS35197H.
- Pareek, V.; Bhargava, A.; Gupta, R.; Jain, N.; Panwar, J. Synthesis and applications of noble metal nanoparticles: a review. Adv. Sci. Eng. Med. 2017, 9, 527-544.
- Amendola, V.; Meneghetti, M. Laser ablation synthesis in solution and size manipulation of noble metal nanoparticles. Phys. Chem. Chem. Phys. 2009, 11, 3805-3821, doi:10.1039/B900654K.
- Cheng, W.; Zhang, W.; Hu, L.; Ding, W.; Wu, F.; Li, J. Etching synthesis of iron oxide nanoparticles for adsorption of arsenic from water. RSC Adv. 2016, 6, 15900-15910, doi:10.1039/C5RA26143K.
- Xu, C.; De, S.; Balu, A.M.; Ojeda, M.; Luque, R. Mechanochemical synthesis of advanced nanomaterials for catalytic applications. Chem. Commun. 2015, 51, 6698-6713.
- Roucoux, A.; Schulz, J.; Patin, H. Reduced transition metal colloids: a novel family of reusable catalysts? Chem. Rev. 2002, 102, 3757-3778.
- Fiévet, F.; Ammar-Merah, S.; Brayner, R.; Chau, F.; Giraud, M.; Mammeri, F.; Peron, J.; Piquemal, J.Y.; Sicard, L.; Viau, G. The polyol process: a unique method for easy access to metal nanoparticles with tailored sizes, shapes and compositions. Chem. Soc. Rev. 2018, 47, 5187-5233, doi:10.1039/C7CS00777A.
- Dong, H.; Chen, Y.C.; Feldmann, C. Polyol synthesis of nanoparticles: status and options regarding metals, oxides, chalcogenides, and non-metal elements. Green Chem. 2015, 17, 4107-4132, doi:10.1039/C5GC00943J.
- An, K.; Alayoglu, S.; Ewers, T.; Somorjai, G.A. Colloid chemistry of nanocatalysts: A molecular view. J. Colloid Interface Sci. 2012, 373, 1-13, doi:https://doi.org/10.1016/j.jcis.2011.10.082.
- Kuhn, J.N.; Tsung, C.-K.; Huang, W.; Somorjai, G.A. Effect of organic capping layers over monodisperse platinum nanoparticles upon activity for ethylene hydrogenation and carbon monoxide oxidation. J. Catal. 2009, 265, 209-215, doi:https://doi.org/10.1016/j.jcat.2009.05.001.
- Noh, J.-H.; Meijboom, R. Catalytic evaluation of dendrimer-templated Pd nanoparticles in the reduction of 4-nitrophenol using Langmuir–Hinshelwood kinetics. Appl. Surf. Sci. 2014, 320, 400-413.
- Hinterwirth, H.; Kappel, S.; Waitz, T.; Prohaska, T.; Lindner, W.; Lämmerhofer, M. Quantifying Thiol Ligand Density of Self-Assembled Monolayers on Gold Nanoparticles by Inductively Coupled Plasma–Mass Spectrometry. ACS Nano 2013, 7, 1129-1136, doi:10.1021/nn306024a.
- Serrano-Maldonado, A.; Martin, E.; Guerrero-Ríos, I. Pyridine-Stabilized Rhodium Nanoparticles in Ionic Liquids as Selective Hydrogenation and Transfer Hydrogenation Catalysts. Eur. J. Inorg. Chem. 2019, 2019, 2863-2870, doi:10.1002/ejic.201900223.
- Gyergyek, S.; Makovec, D.; Drofenik, M. Colloidal stability of oleic- and ricinoleic-acid-coated magnetic nanoparticles in organic solvents. J. Colloid Interface Sci. 2011, 354, 498-505, doi:https://doi.org/10.1016/j.jcis.2010.11.043.
- Singh, A.; Chawla, P.; Jain, S.; Sharma, S.N. Tapping the potential of trioctylphosphine (TOP) in the realization of highly luminescent blue-emitting colloidal indium phosphide (InP) quantum dots. Physica E Low Dimens. Syst. Nanostruct. 2017, 90, 175-182, doi:https://doi.org/10.1016/j.physe.2017.03.029.
- Eriksson, S.; Nylén, U.; Rojas, S.; Boutonnet, M. Preparation of catalysts from microemulsions and their applications in heterogeneous catalysis. Appl. Catal. A Gen. 2004, 265, 207-219.
- Martínez, A.; Prieto, G. The key role of support surface tuning during the preparation of catalysts from reverse micellar-synthesized metal nanoparticles. Catal. Commun. 2007, 8, 1479-1486, doi:https://doi.org/10.1016/j.catcom.2006.12.025.
- Park, J.; Joo, J.; Kwon, S.G.; Jang, Y.; Hyeon, T. Synthesis of Monodisperse Spherical Nanocrystals. Angew. Chem. Int. Ed. 2007, 46, 4630-4660, doi:10.1002/anie.200603148.
- Thanh, N.T.K.; Maclean, N.; Mahiddine, S. Mechanisms of Nucleation and Growth of Nanoparticles in Solution. Chem. Rev. 2014, 114, 7610-7630, doi:10.1021/cr400544s.
- Polte, J. Fundamental growth principles of colloidal metal nanoparticles – a new perspective. CrystEngComm 2015, 17, 6809-6830, doi:10.1039/C5CE01014D.
- Murray, C.B.; Sun, S.; Gaschler, W.; Doyle, H.; Betley, T.A.; Kagan, C.R. Colloidal synthesis of nanocrystals and nanocrystal superlattices. IBM J. Res. Dev. 2001, 45, 47-56.
- Mori, K.; Kumami, A.; Tomonari, M.; Yamashita, H. A pH-induced size controlled deposition of colloidal Ag nanoparticles on alumina support for catalytic application. J. Phys. Chem. C 2009, 113, 16850-16854.
- Pol, V.G.; Srivastava, D.N.; Palchik, O.; Palchik, V.; Slifkin, M.A.; Weiss, A.M.; Gedanken, A. Sonochemical Deposition of Silver Nanoparticles on Silica Spheres. Langmuir 2002, 18, 3352-3357, doi:10.1021/la0155552.
- Doktycz, S.J.; Suslick, K.S. Interparticle collisions driven by ultrasound. Science 1990, 247, 1067-1069.
- Zhang, L.; Cullen, D.A.; Zhai, P.; Ding, K. Adsorption of Colloidal Metal Nanoparticles via Solvent Engineering. ACS Catal. 2020, 10, 2378-2383, doi:10.1021/acscatal.9b05148.
- Tovstun, S.A.; Razumov, V.F. Theory of size-selective precipitation. J. Nanopart. Res. 2016, 19, 8, doi:10.1007/s11051-016-3706-5.
- Quang, D.V.; Lee, J.E.; Kim, J.-K.; Kim, Y.N.; Shao, G.N.; Kim, H.T. A gentle method to graft thiol-functional groups onto silica gel for adsorption of silver ions and immobilization of silver nanoparticles. Powder Technol. 2013, 235, 221-227, doi:https://doi.org/10.1016/j.powtec.2012.10.015.
- Maria Claesson, E.; Philipse, A.P. Thiol-functionalized silica colloids, grains, and membranes for irreversible adsorption of metal(oxide) nanoparticles. Colloids Surf. Physicochem. Eng. Aspects 2007, 297, 46-54, doi:https://doi.org/10.1016/j.colsurfa.2006.10.019.
- Rioux, R.M.; Song, H.; Hoefelmeyer, J.D.; Yang, P.; Somorjai, G.A. High-Surface-Area Catalyst Design: Synthesis, Characterization, and Reaction Studies of Platinum Nanoparticles in Mesoporous SBA-15 Silica. J. Phys. Chem. B 2005, 109, 2192-2202, doi:10.1021/jp048867x.
- Haneveld, J.; Tas, N.R.; Brunets, N.; Jansen, H.V.; Elwenspoek, M. Capillary filling of sub-10nm nanochannels. J. Appl. Phys. 2008, 104, 014309, doi:10.1063/1.2952053.
- Blavo, S.O.; Qayyum, E.; Baldyga, L.M.; Castillo, V.A.; Sanchez, M.D.; Warrington, K.; Barakat, M.A.; Kuhn, J.N. Verification of Organic Capping Agent Removal from Supported Colloidal Synthesized Pt Nanoparticle Catalysts. Top. Catal. 2013, 56, 1835-1842, doi:10.1007/s11244-013-0120-4.
- Huang, W.X.; Hua, Q.; Cao, T. Influence and Removal of Capping Ligands on Catalytic Colloidal Nanoparticles. Catal. Lett. 2014, 144, 1355-1369, doi:10.1007/s10562-014-1306-5.
- Delgado, J.A.; Claver, C.; Castillon, S.; Curulla-Ferre, D.; Ordomsky, V.V.; Godard, C. Effect of polymeric stabilizers on Fischer-Tropsch synthesis catalyzed by cobalt nanoparticles supported on TiO2. J. Mol. Catal. A-Chem. 2016, 417, 43-52, doi:10.1016/j.molcata.2016.02.029.
- He, B.; Zhao, Q.; Zeng, Z.; Wang, X.; Han, S. Effect of hydrothermal reaction time and calcination temperature on properties of Au@CeO2 core–shell catalyst for CO oxidation at low temperature. J. Mater. Sci. 2015, 50, 6339-6348, doi:10.1007/s10853-015-9181-z.
- Tsubota, S.; Nakamura, T.; Tanaka, K.; Haruta, M. Effect of calcination temperature on the catalytic activity of Au colloids mechanically mixed with TiO2 powder for CO oxidation. Catal. Lett. 1998, 56, 131-135, doi:10.1023/A:1019069315071.
- Freund, H.-J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chem. Int. Ed. 2011, 50, 10064-10094, doi:10.1002/anie.201101378.
- Al Soubaihi, R.M.; Saoud, K.M.; Dutta, J. Critical Review of Low-Temperature CO Oxidation and Hysteresis Phenomenon on Heterogeneous Catalysts. Catalysts 2018, 8, 660.
- Xi, K.; Wang, Y.; Jiang, K.; Xie, J.; Zhou, Y.; Lu, H. Support interaction of Pt/CeO2 and Pt/SiC catalysts prepared by nano platinum colloid deposition for CO oxidation. J. Rare Earths 2020, 38, 376-383, doi:https://doi.org/10.1016/j.jre.2019.07.012.
- Ciriminna, R.; Pandarus, V.; Béland, F.; Xu, Y.-J.; Pagliaro, M. Heterogeneously Catalyzed Alcohol Oxidation for the Fine Chemical Industry. Org. Process Res. Dev. 2015, 19, 1554-1558, doi:10.1021/acs.oprd.5b00204.
- Kumar, A.; Kumar, V.P.; Srikanth, A.; Vishwanathan, V.; Chary, K.V.R. Vapor Phase Oxidation of Benzyl Alcohol over Nano Au/SBA-15 Catalysts: Effect of Preparation Methods. Catal. Lett. 2016, 146, 35-46, doi:10.1007/s10562-015-1656-7.
- Saadatjou, N.; Jafari, A.; Sahebdelfar, S. Synthesis and Characterization of Ru/Al2O3 Nanocatalyst for Ammonia Synthesis. Iran J. Chem. Chem. Eng.-Int. Engl. Ed. 2015, 34, 1-9.
- Abashar, M.E.E. Ultra-clean hydrogen production by ammonia decomposition. J. King Saud Univ. Eng. Sci. 2018, 30, 2-11, doi:https://doi.org/10.1016/j.jksues.2016.01.002.
- Choudhary, T.V.; Sivadinarayana, C.; Goodman, D.W. Catalytic ammonia decomposition: COx-free hydrogen production for fuel cell applications. Catal. Lett. 2001, 72, 197-201, doi:10.1023/A:1009023825549.
- Bell, T.E.; Torrente-Murciano, L. H2 Production via Ammonia Decomposition Using Non-Noble Metal Catalysts: A Review. Top. Catal. 2016, 59, 1438-1457, doi:10.1007/s11244-016-0653-4.
- Li, Y.; Wen, J.; Ali, A.M.; Duan, M.; Zhu, W.; Zhang, H.; Chen, C.; Li, Y. Size structure–catalytic performance correlation of supported Ni/MCF-17 catalysts for COx-free hydrogen production. Chem. Commun. 2018, 54, 6364-6367, doi:10.1039/C8CC01884G.
- Zheng, W.; Zhang, J.; Xu, H.; Li, W. NH3 Decomposition Kinetics on Supported Ru Clusters: Morphology and Particle Size Effect. Catal. Lett. 2007, 119, 311-318, doi:10.1007/s10562-007-9237-z.
- Zhang, J.; Xu, H.; Li, W. Kinetic study of NH3 decomposition over Ni nanoparticles: The role of La promoter, structure sensitivity and compensation effect. Appl. Catal. A Gen. 2005, 296, 257-267.
- Mahmoudi, H.; Mahmoudi, M.; Doustdar, O.; Jahangiri, H.; Tsolakis, A.; Gu, S.; LechWyszynski, M. A review of Fischer Tropsch synthesis process, mechanism, surface chemistry and catalyst formulation. Biofuels Eng. 2017, 2, doi:10.1515/bfuel-2017-0002.
- Dalai, A.K.; Davis, B.H. Fischer–Tropsch synthesis: A review of water effects on the performances of unsupported and supported Co catalysts. Appl. Catal. A Gen. 2008, 348, 1-15, doi:https://doi.org/10.1016/j.apcata.2008.06.021.
- Jahangiri, H.; Bennett, J.; Mahjoubi, P.; Wilson, K.; Gu, S. A review of advanced catalyst development for Fischer–Tropsch synthesis of hydrocarbons from biomass derived syn-gas. Catal. Sci. Technol. 2014, 4, 2210-2229, doi:10.1039/C4CY00327F.
- Steynberg, A.P.; Nel, H.G. Clean coal conversion options using Fischer–Tropsch technology. Fuel 2004, 83, 765-770, doi:https://doi.org/10.1016/j.fuel.2003.09.023.
- Melaet, G.; Ralston, W.T.; Li, C.-S.; Alayoglu, S.; An, K.; Musselwhite, N.; Kalkan, B.; Somorjai, G.A. Evidence of Highly Active Cobalt Oxide Catalyst for the Fischer–Tropsch Synthesis and CO2 Hydrogenation. J. Am. Chem. Soc. 2014, 136, 2260-2263, doi:10.1021/ja412447q.
- Delgado, J.A.; Claver, C.; Castillón, S.; Curulla-Ferré, D.; Ordomsky, V.V.; Godard, C. Fischer–Tropsch synthesis catalysed by small TiO2 supported cobalt nanoparticles prepared by sodium borohydride reduction. Appl. Catal. A Gen. 2016, 513, 39-46, doi:https://doi.org/10.1016/j.apcata.2015.12.019.
- Glavee, G.N.; Klabunde, K.J.; Sorensen, C.M.; Hadjipanayis, G.C. Borohydride reduction of cobalt ions in water. Chemistry leading to nanoscale metal, boride, or borate particles. Langmuir 1993, 9, 162-169, doi:10.1021/la00025a034.
- Tan, K.F.; Chang, J.; Borgna, A.; Saeys, M. Effect of boron promotion on the stability of cobalt Fischer–Tropsch catalysts. J. Catal. 2011, 280, 50-59, doi:https://doi.org/10.1016/j.jcat.2011.03.002.
- Saeys, M.; Tan, K.F.; Chang, J.; Borgna, A. Improving the Stability of Cobalt Fischer−Tropsch Catalysts by Boron Promotion. Ind. Eng. Chem. Res. 2010, 49, 11098-11100, doi:10.1021/ie100523u.
- Krans, N.A.; Weber, J.L.; Van den Bosch, W.; Zecevic, J.; de Jongh, P.E.; de Jong, K.P. Influence of Promotion on the Growth of Anchored Colloidal Iron Oxide Nanoparticles during Synthesis Gas Conversion. ACS Catal. 2020, 10, 1913-1922, doi:10.1021/acscatal.9b04380.
- Xie, J.; Torres Galvis, H.M.; Koeken, A.C.J.; Kirilin, A.; Dugulan, A.I.; Ruitenbeek, M.; de Jong, K.P. Size and Promoter Effects on Stability of Carbon-Nanofiber-Supported Iron-Based Fischer–Tropsch Catalysts. ACS Catal. 2016, 6, 4017-4024, doi:10.1021/acscatal.6b00321.
- van Hoof, A.J.F.; Michel-Ligthart, D.A.J.; Friedrich, H.; Hensen, E.J.M. The Influence and Removability of Colloidal Capping Agents on Carbon Monoxide Hydrogenation by Zirconia-Supported Rhodium Nanoparticles. Chemcatchem 2017, 9, 1018-1024, doi:10.1002/cctc.201601378.
- Lam, E.; Larmier, K.; Wolf, P.; Tada, S.; Safonova, O.V.; Copéret, C. Isolated Zr Surface Sites on Silica Promote Hydrogenation of CO2 to CH3OH in Supported Cu Catalysts. J. Am. Chem. Soc. 2018, 140, 10530-10535, doi:10.1021/jacs.8b05595.
- Singh, A.K.; Xu, Q. Synergistic Catalysis over Bimetallic Alloy Nanoparticles. ChemCatChem 2013, 5, 652-676, doi:10.1002/cctc.201200591.
- Ferrando, R.; Jellinek, J.; Johnston, R.L. Nanoalloys: From Theory to Applications of Alloy Clusters and Nanoparticles. Chem. Rev. 2008, 108, 845-910, doi:10.1021/cr040090g.
- Jiang, H.-L.; Xu, Q. Recent progress in synergistic catalysis over heterometallic nanoparticles. J. Mater. Chem. 2011, 21, 13705-13725, doi:10.1039/C1JM12020D.
- Wang, D.; Li, Y. Bimetallic Nanocrystals: Liquid-Phase Synthesis and Catalytic Applications. Adv. Mater. 2011, 23, 1044-1060, doi:10.1002/adma.201003695.
- Destro, P. Colloidal Nanoparticles for Heterogeneous Catalysis; Springer: 2018.
- Destro, P.; Marras, S.; Manna, L.; Colombo, M.; Zanchet, D. AuCu alloy nanoparticles supported on SiO2: Impact of redox pretreatments in the catalyst performance in CO oxidation. Catal. Today 2017, 282, 105-110, doi:https://doi.org/10.1016/j.cattod.2016.08.003.
- Destro, P.; Kokumai, T.M.; Scarpellini, A.; Pasquale, L.; Manna, L.; Colombo, M.; Zanchet, D. The Crucial Role of the Support in the Transformations of Bimetallic Nanoparticles and Catalytic Performance. ACS Catal. 2018, 8, 1031-1037, doi:10.1021/acscatal.7b03685.
- Najafishirtari, S.; Brescia, R.; Guardia, P.; Marras, S.; Manna, L.; Colombo, M. Nanoscale Transformations of Alumina-Supported AuCu Ordered Phase Nanocrystals and Their Activity in CO Oxidation. ACS Catal. 2015, 5, 2154-2163, doi:10.1021/cs501923x.
- Zaytsev, S.Y.; Plyusnin, P.E.; Slavinskaya, E.M.; Shubin, Y.V. Synthesis of bimetallic nanocompositions AuxPd1-x/γ-Al2O3 for catalytic CO oxidation. J. Nanopart. Res. 2017, 19, 367, doi:10.1007/s11051-017-4061-x.
- Nagy, G.; Benkó, T.; Borkó, L.; Csay, T.; Horváth, A.; Frey, K.; Beck, A. Bimetallic Au–Ag/SiO2 catalysts: comparison in glucose, benzyl alcohol and CO oxidation reactions. React. Kinet. Mech. Catal. 2015, 115, 45-65, doi:10.1007/s11144-015-0835-2.
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for combustion of methane and lower alkanes. Appl. Catal. A Gen. 2002, 234, 1-23, doi:https://doi.org/10.1016/S0926-860X(02)00231-4.
- Xiong, H.; Wiebenga, M.H.; Carrillo, C.; Gaudet, J.R.; Pham, H.N.; Kunwar, D.; Oh, S.H.; Qi, G.; Kim, C.H.; Datye, A.K. Design considerations for low-temperature hydrocarbon oxidation reactions on Pd based catalysts. Appl. Catal. B Environ. 2018, 236, 436-444, doi:https://doi.org/10.1016/j.apcatb.2018.05.049.
- Nie, H.; Howe, J.Y.; Lachkov, P.T.; Chin, Y.-H.C. Chemical and Structural Dynamics of Nanostructures in Bimetallic Pt–Pd Catalysts, Their Inhomogeneity, and Their Roles in Methane Oxidation. ACS Catal. 2019, 9, 5445-5461, doi:10.1021/acscatal.9b00485.
- Nassiri, H.; Lee, K.-E.; Hu, Y.; Hayes, R.E.; Scott, R.W.J.; Semagina, N. Platinum Inhibits Low-Temperature Dry Lean Methane Combustion through Palladium Reduction in Pd−Pt/Al2O3: An In Situ X-ray Absorption Study. Chemphyschem 2017, 18, 238-244, doi:10.1002/cphc.201600993.
- Persson, K.; Jansson, K.; Järås, S.G. Characterisation and microstructure of Pd and bimetallic Pd–Pt catalysts during methane oxidation. J. Catal. 2007, 245, 401-414, doi:https://doi.org/10.1016/j.jcat.2006.10.029.
- Qu, P.; Wang, S.; Hu, W.; Wu, Y.; Chen, J.; Zhang, G.; Shen, P.; Chen, Y.; Zhong, L. A novel strategy to design PtPd bimetallic catalysts for efficient methane combustion. Catal. Commun. 2020, 135, 105900, doi:https://doi.org/10.1016/j.catcom.2019.105900.
- Cano, L.A.; Garcia Blanco, A.A.; Lener, G.; Marchetti, S.G.; Sapag, K. Effect of the support and promoters in Fischer-Tropsch synthesis using supported Fe catalysts. Catal. Today 2017, 282, 204-213, doi:https://doi.org/10.1016/j.cattod.2016.06.054.
- Ismail, A.S.M.; Casavola, M.; Liu, B.; Gloter, A.; van Deelen, T.W.; Versluijs, M.; Meeldijk, J.D.; Stéphan, O.; de Jong, K.P.; de Groot, F.M.F. Atomic-Scale Investigation of the Structural and Electronic Properties of Cobalt–Iron Bimetallic Fischer–Tropsch Catalysts. ACS Catal. 2019, 9, 7998-8011, doi:10.1021/acscatal.8b04334.
- Dad, M.; Lancee, R.J.; Janse van Vuuren, M.; van de Loosdrecht, J.; Niemantsverdriet, J.W.H.; Fredriksson, H.O.A. SiO2-supported Fe & FeMn colloids—Fischer-Tropsch synthesis on 3D model catalysts. Appl. Catal. A Gen. 2017, 537, 83-92, doi:https://doi.org/10.1016/j.apcata.2017.02.023.
- Jiang, N.; Yang, G.; Zhang, X.; Wang, L.; Shi, C.; Tsubaki, N. A novel silicalite-1 zeolite shell encapsulated iron-based catalyst for controlling synthesis of light alkenes from syngas. Catal. Commun. 2011, 12, 951-954, doi:https://doi.org/10.1016/j.catcom.2011.02.021.
- Yang, G.; Tan, Y.; Han, Y.; Qiu, J.; Tsubaki, N. Increasing the shell thickness by controlling the core size of zeolite capsule catalyst: Application in iso-paraffin direct synthesis. Catal. Commun. 2008, 9, 2520-2524, doi:https://doi.org/10.1016/j.catcom.2008.07.006.
- Zeng, B.; Hou, B.; Jia, L.; Wang, J.; Chen, C.; Li, D.; Sun, Y. The intrinsic effects of shell thickness on the Fischer–Tropsch synthesis over core–shell structured catalysts. Catal. Sci. Technol. 2013, 3, 3250-3255, doi:10.1039/C3CY00665D.
- Haghtalab, A.; Mosayebi, A. Co@Ru nanoparticle with core–shell structure supported over γ-Al2O3 for Fischer–Tropsch synthesis. Int. J. Hydrogen Energy 2014, 39, 18882-18893, doi:https://doi.org/10.1016/j.ijhydene.2014.09.074.
- Semelsberger, T.A.; Borup, R.L.; Greene, H.L. Dimethyl ether (DME) as an alternative fuel. J. Power Sources 2006, 156, 497-511.
- Anggarani, R.; Wibowo, C.S.; Rulianto, D. Application of dimethyl ether as LPG substitution for household stove. Energy Procedia 2014, 47, 227-234.
- Olah, G.A.; Goeppert, A.; Prakash, G.S. Chemical recycling of carbon dioxide to methanol and dimethyl ether: from greenhouse gas to renewable, environmentally carbon neutral fuels and synthetic hydrocarbons. J. Org. Chem. 2009, 74, 487-498.
- French, S.; Sokol, A.; Bromley, S.; Catlow, C.; Sherwood, P. Identification and characterization of active sites and their catalytic processes—the Cu/ZnO methanol catalyst. Top. Catal. 2003, 24, 161-172.
- Stiefel, M.; Ahmad, R.; Arnold, U.; Döring, M. Direct synthesis of dimethyl ether from carbon-monoxide-rich synthesis gas: Influence of dehydration catalysts and operating conditions. Fuel Process. Technol. 2011, 92, 1466-1474, doi:https://doi.org/10.1016/j.fuproc.2011.03.007.
- Zhang, Q.; Li, X.; Asami, K.; Asaoka, S.; Fujimoto, K. Synthesis of LPG from synthesis gas. Fuel Process. Technol. 2004, 85, 1139-1150, doi:https://doi.org/10.1016/j.fuproc.2003.10.016.
- Gentzen, M.; Habicht, W.; Doronkin, D.E.; Grunwaldt, J.D.; Sauer, J.; Behrens, S. Bifunctional hybrid catalysts derived from Cu/Zn-based nanoparticles for single-step dimethyl ether synthesis. Catal. Sci. Technol. 2016, 6, 1054-1063, doi:10.1039/C5CY01043H.
- Gentzen, M.; Doronkin, D.E.; Sheppard, T.L.; Grunwaldt, J.D.; Sauer, J.; Behrens, S. Bifunctional catalysts based on colloidal Cu/Zn nanoparticles for the direct conversion of synthesis gas to dimethyl ether and hydrocarbons. Appl. Catal. A Gen. 2018, 557, 99-107, doi:https://doi.org/10.1016/j.apcata.2018.03.008.
- Lebarbier, V.M.; Dagle, R.A.; Kovarik, L.; Lizarazo-Adarme, J.A.; King, D.L.; Palo, D.R. Synthesis of methanol and dimethyl ether from syngas over Pd/ZnO/Al 2 O 3 catalysts. Catal. Sci. Technol. 2012, 2, 2116-2127.
- Bahruji, H.; Bowker, M.; Jones, W.; Hayward, J.; Esquius, J.R.; Morgan, D.J.; Hutchings, G.J. PdZn catalysts for CO2 hydrogenation to methanol using chemical vapour impregnation (CVI). Faraday Discuss. 2017, 197, 309-324.
- Gentzen, M.; Doronkin, D.E.; Sheppard, T.L.; Zimina, A.; Li, H.; Jelic, J.; Studt, F.; Grunwaldt, J.-D.; Sauer, J.; Behrens, S. Supported Intermetallic PdZn Nanoparticles as Bifunctional Catalysts for the Direct Synthesis of Dimethyl Ether from CO-Rich Synthesis Gas. Angew. Chem. Int. Ed. 2019, 58, 15655-15659, doi:10.1002/anie.201906256.
- Rajesh Kumar, B.; Saravanan, S. Use of higher alcohol biofuels in diesel engines: A review. Renew. Sust. Energ. Rev. 2016, 60, 84-115, doi:https://doi.org/10.1016/j.rser.2016.01.085.
- Lu, Y.; Cao, B.; Yu, F.; Liu, J.; Bao, Z.; Gao, J. High Selectivity Higher Alcohols Synthesis from Syngas over Three-Dimensionally Ordered Macroporous Cu-Fe Catalysts. ChemCatChem 2014, 6, 473-478, doi:10.1002/cctc.201300749.
- Gao, W.; Zhao, Y.; Liu, J.; Huang, Q.; He, S.; Li, C.; Zhao, J.; Wei, M. Catalytic conversion of syngas to mixed alcohols over CuFe-based catalysts derived from layered double hydroxides. Catal. Sci. Technol. 2013, 3, 1324-1332, doi:10.1039/C3CY00025G.
- Xiao, K.; Bao, Z.; Qi, X.; Wang, X.; Zhong, L.; Fang, K.; Lin, M.; Sun, Y. Structural evolution of CuFe bimetallic nanoparticles for higher alcohol synthesis. J. Mol. Catal. A: Chem. 2013, 378, 319-325, doi:https://doi.org/10.1016/j.molcata.2013.07.006.
- Xiao, K.; Qi, X.; Bao, Z.; Wang, X.; Zhong, L.; Fang, K.; Lin, M.; Sun, Y. CuFe, CuCo and CuNi nanoparticles as catalysts for higher alcohol synthesis from syngas: a comparative study. Catal. Sci. Technol. 2013, 3, 1591-1602, doi:10.1039/C3CY00063J.
- He, S.; Wang, W.; Shen, Z.; Li, G.; Kang, J.; Liu, Z.; Wang, G.-C.; Zhang, Q.; Wang, Y. Carbon nanotube-supported bimetallic Cu-Fe catalysts for syngas conversion to higher alcohols. Mol. Catal. 2019, 479, 110610, doi:https://doi.org/10.1016/j.mcat.2019.110610.
- Aitbekova, A.; Goodman, E.D.; Wu, L.; Boubnov, A.; Hoffman, A.S.; Genc, A.; Cheng, H.; Casalena, L.; Bare, S.R.; Cargnello, M. Engineering of Ruthenium–Iron Oxide Colloidal Heterostructures: Improved Yields in CO2 Hydrogenation to Hydrocarbons. Angew. Chem. Int. Ed. 2019, 58, 17451-17457, doi:10.1002/anie.201910579.
- McCue, A.J.; Anderson, J.A. Recent advances in selective acetylene hydrogenation using palladium containing catalysts. Front. Chem. Sci. Eng. 2015, 9, 142-153, doi:10.1007/s11705-015-1516-4.
- Bruno, J.E.; Dwarica, N.S.; Whittaker, T.N.; Hand, E.R.; Guzman, C.S.; Dasgupta, A.; Chen, Z.; Rioux, R.M.; Chandler, B.D. Supported Ni–Au Colloid Precursors for Active, Selective, and Stable Alkyne Partial Hydrogenation Catalysts. ACS Catal. 2020, 10, 2565-2580, doi:10.1021/acscatal.9b05402.
- Nawaz, Z. Light alkane dehydrogenation to light olefin technologies: a comprehensive review. Rev. Chem. Eng. 2015, 31, 413-436.
- Iglesias-Juez, A.; Beale, A.M.; Maaijen, K.; Weng, T.C.; Glatzel, P.; Weckhuysen, B.M. A combined in situ time-resolved UV–Vis, Raman and high-energy resolution X-ray absorption spectroscopy study on the deactivation behavior of Pt and PtSn propane dehydrogenation catalysts under industrial reaction conditions. J. Catal. 2010, 276, 268-279, doi:https://doi.org/10.1016/j.jcat.2010.09.018.
- Bariås, O.A.; Holmen, A.; Blekkan, E.A. Propane dehydrogenation over supported Pt and Pt–Sn catalysts: catalyst preparation, characterization, and activity measurements. J. Catal. 1996, 158, 1-12.
- Yu, C.; Ge, Q.; Xu, H.; Li, W. Effects of Ce addition on the Pt-Sn/γ-Al2O3 catalyst for propane dehydrogenation to propylene. Appl. Catal. A Gen. 2006, 315, 58-67.
- Zhang, Y.; Zhou, Y.; Qiu, A.; Wang, Y.; Xu, Y.; Wu, P. Propane dehydrogenation on PtSn/ZSM-5 catalyst: Effect of tin as a promoter. Catal. Commun. 2006, 7, 860-866.
- Lobera, M.; Tellez, C.; Herguido, J.; Menéndez, M. Transient kinetic modelling of propane dehydrogenation over a Pt–Sn–K/Al2O3 catalyst. Appl. Catal. A Gen. 2008, 349, 156-164.
- Vu, B.K.; Song, M.B.; Ahn, I.Y.; Suh, Y.-W.; Suh, D.J.; Kim, W.-I.; Koh, H.-L.; Choi, Y.G.; Shin, E.W. Pt–Sn alloy phases and coke mobility over Pt–Sn/Al2O3 and Pt–Sn/ZnAl2O4 catalysts for propane dehydrogenation. Appl. Catal. A Gen. 2011, 400, 25-33.
- Zhu, H.; Anjum, D.H.; Wang, Q.; Abou-Hamad, E.; Emsley, L.; Dong, H.; Laveille, P.; Li, L.; Samal, A.K.; Basset, J.-M. Sn surface-enriched Pt–Sn bimetallic nanoparticles as a selective and stable catalyst for propane dehydrogenation. J. Catal. 2014, 320, 52-62.
- Kaylor, N.; Davis, R.J. Propane dehydrogenation over supported Pt-Sn nanoparticles. J. Catal. 2018, 367, 181-193, doi:https://doi.org/10.1016/j.jcat.2018.09.006.
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Marin, G.B. Enhanced Carbon-Resistant Dry Reforming Fe-Ni Catalyst: Role of Fe. ACS Catal. 2015, 5, 3028-3039, doi:10.1021/acscatal.5b00357.
- Theofanidis, S.A.; Galvita, V.V.; Sabbe, M.; Poelman, H.; Detavernier, C.; Marin, G.B. Controlling the stability of a Fe–Ni reforming catalyst: Structural organization of the active components. Appl. Catal. B Environ. 2017, 209, 405-416, doi:https://doi.org/10.1016/j.apcatb.2017.03.025.
- de Lima, S.M.; Assaf, J.M. Ni–Fe Catalysts Based on Perovskite-type Oxides for Dry Reforming of Methane to Syngas. Catal. Lett. 2006, 108, 63-70, doi:10.1007/s10562-006-0026-x.
- Medeiros, R.L.B.A.; Macedo, H.P.; Melo, V.R.M.; Oliveira, Â.A.S.; Barros, J.M.F.; Melo, M.A.F.; Melo, D.M.A. Ni supported on Fe-doped MgAl2O4 for dry reforming of methane: Use of factorial design to optimize H2 yield. Int. J. Hydrogen Energy 2016, 41, 14047-14057, doi:https://doi.org/10.1016/j.ijhydene.2016.06.246.
- Theofanidis, S.A.; Batchu, R.; Galvita, V.V.; Poelman, H.; Marin, G.B. Carbon gasification from Fe–Ni catalysts after methane dry reforming. Appl. Catal. B Environ. 2016, 185, 42-55, doi:https://doi.org/10.1016/j.apcatb.2015.12.006.
- Theofanidis, S.A.; Galvita, V.V.; Poelman, H.; Dharanipragada, N.V.R.A.; Longo, A.; Meledina, M.; Van Tendeloo, G.; Detavernier, C.; Marin, G.B. Fe-Containing Magnesium Aluminate Support for Stability and Carbon Control during Methane Reforming. ACS Catal. 2018, 8, 5983-5995, doi:10.1021/acscatal.8b01039.
- Kim, S.M.; Abdala, P.M.; Margossian, T.; Hosseini, D.; Foppa, L.; Armutlulu, A.; van Beek, W.; Comas-Vives, A.; Copéret, C.; Müller, C. Cooperativity and Dynamics Increase the Performance of NiFe Dry Reforming Catalysts. J. Am. Chem. Soc. 2017, 139, 1937-1949, doi:10.1021/jacs.6b11487.
- Theofanidis, S.A.; Poelman, H.; Marin, G.B.; Galvita, V.V. Chapter 6 - How Does the Surface Structure of Ni-Fe Nanoalloys Control Carbon Formation During Methane Steam/Dry Reforming? In Advanced Nanomaterials for Catalysis and Energy, Sadykov, V.A., Ed. Elsevier: Amsterdam, Netherlands, 2019; https://doi.org/10.1016/B978-0-12-814807-5.00006-1pp. 177-225.
- Margossian, T.; Larmier, K.; Kim, S.M.; Krumeich, F.; Müller, C.; Copéret, C. Supported Bimetallic NiFe Nanoparticles through Colloid Synthesis for Improved Dry Reforming Performance. ACS Catal. 2017, 7, 6942-6948, doi:10.1021/acscatal.7b02091.
- Li, Z.W.; Wang, Z.G.; Kawi, S. Sintering and Coke Resistant Core/Yolk Shell Catalyst for Hydrocarbon Reforming. Chemcatchem 2019, 11, 202-224, doi:10.1002/cctc.201801266.
- Park, J.C.; Song, H. Metal@Silica Yolk-Shell Nanostructures as Versatile Bifunctional Nanocatalysts. Nano Res. 2011, 4, 33-49, doi:10.1007/s12274-010-0039-z.
- Zhang, Q.; Zhang, T.; Ge, J.; Yin, Y. Permeable Silica Shell through Surface-Protected Etching. Nano Lett. 2008, 8, 2867-2871, doi:10.1021/nl8016187.
- Zhang, Q.; Lee, I.; Ge, J.; Zaera, F.; Yin, Y. Surface-Protected Etching of Mesoporous Oxide Shells for the Stabilization of Metal Nanocatalysts. Adv. Funct. Mater. 2010, 20, 2201-2214, doi:10.1002/adfm.201000428.
- Galeano, C.; Güttel, R.; Paul, M.; Arnal, P.; Lu, A.-H.; Schüth, F. Yolk-Shell Gold Nanoparticles as Model Materials for Support-Effect Studies in Heterogeneous Catalysis: Au, @C and Au, @ZrO2 for CO Oxidation as an Example. Chem. Eur. 2011, 17, 8434-8439, doi:10.1002/chem.201100318.
- Prieto, G.; Tüysüz, H.; Duyckaerts, N.; Knossalla, J.; Wang, G.-H.; Schüth, F. Hollow Nano- and Microstructures as Catalysts. Chem. Rev. 2016, 116, 14056-14119, doi:10.1021/acs.chemrev.6b00374.
- Bakhmutsky, K.; Wieder, N.L.; Cargnello, M.; Galloway, B.; Fornasiero, P.; Gorte, R.J. A Versatile Route to Core-Shell Catalysts: Synthesis of Dispersible M@Oxide (M=Pd, Pt; Oxide=TiO2, ZrO2) Nanostructures by Self-Assembly. Chemsuschem 2012, 5, 140-148, doi:10.1002/cssc.201100491.
- Seo, C.Y.; Chen, X.; Sun, K.; Allard, L.F.; Fisher, G.B.; Schwank, J.W. Palladium redispersion at high temperature within the Pd@SiO2 core@shell structure. Catal. Commun. 2018, 108, 73-76, doi:https://doi.org/10.1016/j.catcom.2018.01.027.
- Shirman, E.; Shirman, T.; Shneidman, A.V.; Grinthal, A.; Phillips, K.R.; Whelan, H.; Bulger, E.; Abramovitch, M.; Patil, J.; Nevarez, R., et al. Modular Design of Advanced Catalytic Materials Using Hybrid Organic–Inorganic Raspberry Particles. Adv. Funct. Mater. 2018, 28, 1704559, doi:10.1002/adfm.201704559.
- Shirman, T.; Lattimer, J.; Luneau, M.; Shirman, E.; Reece, C.; Aizenberg, M.; Madix, R.J.; Aizenberg, J.; Friend, C.M. New Architectures for Designed Catalysts: Selective Oxidation using AgAu Nanoparticles on Colloid-Templated Silica. Chem.-Eur. J. 2018, 24, 1833-1837, doi:10.1002/chem.201704552.
- Luneau, M.; Shirman, T.; Filie, A.; Timoshenko, J.; Chen, W.; Trimpalis, A.; Flytzani-Stephanopoulos, M.; Kaxiras, E.; Frenkel, A.I.; Aizenberg, J., et al. Dilute Pd/Au Alloy Nanoparticles Embedded in Colloid-Templated Porous SiO2: Stable Au-Based Oxidation Catalysts. Chem. Mater. 2019, 31, 5759-5768, doi:10.1021/acs.chemmater.9b01779.
- Kim, S.; Lee, S.; Jung, W. Sintering Resistance of Pt@SiO2 Core-Shell Catalyst. Chemcatchem 2019, 11, 4653-4659, doi:10.1002/cctc.201900934.
- Habibi, A.H.; Hayes, R.E.; Semagina, N. Evaluation of hydrothermal stability of encapsulated PdPt@SiO2 catalyst for lean CH4 combustion. Appl. Catal. A Gen. 2018, 556, 129-136, doi:https://doi.org/10.1016/j.apcata.2018.02.034.
- Pei, W.; Liu, Y.; Deng, J.; Zhang, K.; Hou, Z.; Zhao, X.; Dai, H. Partially embedding Pt nanoparticles in the skeleton of 3DOM Mn2O3: An effective strategy for enhancing catalytic stability in toluene combustion. Appl. Catal. B Environ. 2019, 256, 117814, doi:https://doi.org/10.1016/j.apcatb.2019.117814.
- Sadakane, M.; Ueda, W. Three-Dimensionally Ordered Macroporous (3DOM) Perovskite Mixed Metal Oxides. In Perovskites and Related Mixed Oxides, Granger, P., Parvulescu, V.I., Prellier, W., Eds. Wiley: Weinheim, Germany, 2015; doi:10.1002/9783527686605.ch06pp. 113-142.
- Bezemer, G.L.; Bitter, J.H.; Kuipers, H.P.C.E.; Oosterbeek, H.; Holewijn, J.E.; Xu, X.; Kapteijn, F.; van Dillen, A.J.; de Jong, K.P. Cobalt Particle Size Effects in the Fischer−Tropsch Reaction Studied with Carbon Nanofiber Supported Catalysts. J. Am. Chem. Soc. 2006, 128, 3956-3964, doi:10.1021/ja058282w.
- Wielers, A.F.H.; Kock, A.J.H.M.; Hop, C.E.C.A.; Geus, J.W.; van Der Kraan, A.M. The reduction behavior of silica-supported and alumina-supported iron catalysts: A Mössbauer and infrared spectroscopic study. J. Catal. 1989, 117, 1-18, doi:https://doi.org/10.1016/0021-9517(89)90216-9.
- Ni, Z.; Qin, H.; Kang, S.; Bai, J.; Wang, Z.; Li, Y.; Zheng, Z.; Li, X. Effect of graphitic carbon modification on the catalytic performance of Fe@SiO2-GC catalysts for forming lower olefins via Fischer-Tropsch synthesis. J. Colloid Interface Sci. 2018, 516, 16-22, doi:https://doi.org/10.1016/j.jcis.2018.01.017.
- Huang, C.; Zhang, M.W.; Zhu, C.; Mu, X.L.; Zhang, K.; Zhong, L.S.; Fang, K.G.; Wu, M.H. Fabrication of Highly Stable SiO2 Encapsulated Multiple CuFe Nanoparticles for Higher Alcohols Synthesis via CO Hydrogenation. Catal. Lett. 2018, 148, 1080-1092, doi:10.1007/s10562-018-2329-0.
- Ilsemann, J.; Strass-Eifert, A.; Friedland, J.; Kiewidt, L.; Thoming, J.; Baumer, M.; Guttel, R. Cobalt@Silica Core-Shell Catalysts for Hydrogenation of CO/CO2 Mixtures to Methane. Chemcatchem 2019, 11, 4884-4893, doi:10.1002/cctc.201900916.
- Luneau, M.; Shirman, T.; Foucher, A.C.; Duanmu, K.; Verbart, D.M.A.; Sautet, P.; Stach, E.A.; Aizenberg, J.; Madix, R.J.; Friend, C.M. Achieving High Selectivity for Alkyne Hydrogenation at High Conversions with Compositionally Optimized PdAu Nanoparticle Catalysts in Raspberry Colloid-Templated SiO2. ACS Catal. 2020, 10, 441-450, doi:10.1021/acscatal.9b04243.
- Zhang, Y.; Diao, W.; Williams, C.T.; Monnier, J.R. Selective hydrogenation of acetylene in excess ethylene using Ag- and Au–Pd/SiO2 bimetallic catalysts prepared by electroless deposition. Appl. Catal. A Gen. 2014, 469, 419-426, doi:https://doi.org/10.1016/j.apcata.2013.10.024.
- Li, M.; Shen, J. Microcalorimetric and infrared spectroscopic studies of CO and C2H4 adsorption on Pd/SiO2 and Pd–Ag/SiO2 catalysts. Mater. Chem. Phys. 2001, 68, 204-209.
- Teschner, D.; Borsodi, J.; Wootsch, A.; Révay, Z.; Hävecker, M.; Knop-Gericke, A.; Jackson, S.D.; Schlögl, R. The roles of subsurface carbon and hydrogen in palladium-catalyzed alkyne hydrogenation. Science 2008, 320, 86-89.
- Zhao, S.; Li, Y.; Liu, D.; Liu, J.; Liu, Y.-M.; Zakharov, D.N.; Wu, Q.; Orlov, A.; Gewirth, A.A.; Stach, E.A. Multimodal study of the speciations and activities of supported Pd catalysts during the hydrogenation of ethylene. J. Phys. Chem. C 2017, 121, 18962-18972.
- Yang, H.; Gao, P.; Zhang, C.; Zhong, L.; Li, X.; Wang, S.; Wang, H.; Wei, W.; Sun, Y. Core–shell structured Cu@m-SiO2 and Cu/ZnO@m-SiO2 catalysts for methanol synthesis from CO2 hydrogenation. Catal. Commun. 2016, 84, 56-60, doi:https://doi.org/10.1016/j.catcom.2016.06.010.
- Shi, Z.S.; Tan, Q.Q.; Wu, D.F. A novel Core-Shell structured CuIn@SiO2 catalyst for CO2 hydrogenation to methanol. AIChE J. 2019, 65, 1047-1058, doi:10.1002/aic.16490.
- Kawi, S.; Kathiraser, Y.; Ni, J.; Oemar, U.; Li, Z.; Saw, E.T. Progress in Synthesis of Highly Active and Stable Nickel-Based Catalysts for Carbon Dioxide Reforming of Methane. ChemSusChem 2015, 8, 3556-3575, doi:10.1002/cssc.201500390.
- Wang, F.; Han, B.; Zhang, L.; Xu, L.; Yu, H.; Shi, W. CO2 reforming with methane over small-sized Ni@SiO2 catalysts with unique features of sintering-free and low carbon. Appl. Catal. B Environ. 2018, 235, 26-35, doi:https://doi.org/10.1016/j.apcatb.2018.04.069.
- Zhang, L.; Wang, F.; Zhu, J.; Han, B.; Fan, W.; Zhao, L.; Cai, W.; Li, Z.; Xu, L.; Yu, H., et al. CO2 reforming with methane reaction over Ni@SiO2 catalysts coupled by size effect and metal-support interaction. Fuel 2019, 256, 115954, doi:https://doi.org/10.1016/j.fuel.2019.115954.
- Han, B.; Wang, F.; Zhang, L.; Wang, Y.; Fan, W.; Xu, L.; Yu, H.; Li, Z. Syngas production from methane steam reforming and dry reforming reactions over sintering-resistant Ni@SiO2 catalyst. Res. Chem. Intermed. 2020, 46, 1735-1748, doi:10.1007/s11164-019-04060-3.
- LeValley, T.L.; Richard, A.R.; Fan, M. The progress in water gas shift and steam reforming hydrogen production technologies – A review. Int. J. Hydrogen Energy 2014, 39, 16983-17000, doi:https://doi.org/10.1016/j.ijhydene.2014.08.041.
- Hwang, K.-R.; Lee, C.-B.; Park, J.-S. Advanced nickel metal catalyst for water–gas shift reaction. J. Power Sources 2011, 196, 1349-1352, doi:https://doi.org/10.1016/j.jpowsour.2010.08.084.
- Panagiotopoulou, P.; Kondarides, D.I. Effect of the nature of the support on the catalytic performance of noble metal catalysts for the water–gas shift reaction. Catal. Today 2006, 112, 49-52, doi:https://doi.org/10.1016/j.cattod.2005.11.026.
- Ashok, J.; Wai, M.H.; Kawi, S. Nickel-based Catalysts for High-temperature Water Gas Shift Reaction-Methane Suppression. ChemCatChem 2018, 10, 3927-3942, doi:10.1002/cctc.201800031.
- Gao, L.; Ta, N.; Dong, J.; Song, T.; Chen, S.; Fu, Q. Facile Transformation of Ni-based Colloids into Highly Stable Nanocatalysts Embedded within h-BN for the Water-Gas Shift Reaction. ChemCatChem 2020, 12, 1556-1561, doi:10.1002/cctc.201902263.
- Fu, L.; Cheng, G.; Luo, W. Colloidal synthesis of monodisperse trimetallic IrNiFe nanoparticles as highly active bifunctional electrocatalysts for acidic overall water splitting. J. Mater. Chem. A 2017, 5, 24836-24841, doi:10.1039/C7TA08982A.
- Li, J.; Luo, Z.; He, F.; Zuo, Y.; Zhang, C.; Liu, J.; Yu, X.; Du, R.; Zhang, T.; Infante-Carrió, M.F., et al. Colloidal Ni–Co–Sn nanoparticles as efficient electrocatalysts for the methanol oxidation reaction. J. Mater. Chem. A 2018, 6, 22915-22924, doi:10.1039/C8TA08242A.
- Dong, X.; Zheng, P.; Zheng, A.-G.; Li, H.-F.; Xia, G.-F.; Li, M.-F.; Zheng, R.-Y.; Xu, B.-Q. Noble-metal efficient Pt-Ir-Co/SiO2 catalyst for selective hydrogenolytic ring opening of methylcyclopentane. Catal. Today 2018, 316, 162-170, doi:https://doi.org/10.1016/j.cattod.2018.03.059.
- Müller, P.; Hermans, I. Applications of Modulation Excitation Spectroscopy in Heterogeneous Catalysis. Ind. Eng. Chem. Res. 2017, 56, 1123-1136, doi:10.1021/acs.iecr.6b04855.
- Ferri, D.; Newton, M.A.; Nachtegaal, M. Modulation Excitation X-Ray Absorption Spectroscopy to Probe Surface Species on Heterogeneous Catalysts. Top. Catal. 2011, 54, 1070–1078, doi:10.1007/s11244-011-9727-5.
- Gaur, A.; Hartmann Dabros, T.M.; Høj, M.; Boubnov, A.; Prüssmann, T.; Jelic, J.; Studt, F.; Jensen, A.D.; Grunwaldt, J.-D. Probing the Active Sites of MoS2 Based Hydrotreating Catalysts Using Modulation Excitation Spectroscopy. ACS Catal. 2019, 9, 2568-2579, doi:10.1021/acscatal.8b04778.
- Aguirre, A.; Collins, S.E. Selective detection of reaction intermediates using concentration-modulation excitation DRIFT spectroscopy. Catal. Today 2013, 205, 34-40, doi:https://doi.org/10.1016/j.cattod.2012.08.020.
- Nuguid, R.J.G.; Ferri, D.; Marberger, A.; Nachtegaal, M.; Kröcher, O. Modulated Excitation Raman Spectroscopy of V2O5/TiO2: Mechanistic Insights into the Selective Catalytic Reduction of NO with NH3. ACS Catal. 2019, 9, 6814-6820, doi:10.1021/acscatal.9b01514.
- Ferri, D.; Newton, M.A.; Di Michiel, M.; Chiarello, G.L.; Yoon, S.; Lu, Y.; Andrieux, J. Revealing the Dynamic Structure of Complex Solid Catalysts Using Modulated Excitation X‐ray Diffraction. Angew. Chem. Int. Ed. 2014, 53, 8890-8894.
- Morgan, K.; Maguire, N.; Fushimi, R.; Gleaves, J.T.; Goguet, A.; Harold, M.P.; Kondratenko, E.V.; Menon, U.; Schuurman, Y.; Yablonsky, G.S. Forty years of temporal analysis of products. Catal. Sci. Technol. 2017, 7, 2416-2439, doi:10.1039/C7CY00678K.
- Ledesma, C.; Yang, J.; Chen, D.; Holmen, A. Recent Approaches in Mechanistic and Kinetic Studies of Catalytic Reactions Using SSITKA Technique. ACS Catal. 2014, 4, 4527-4547, doi:10.1021/cs501264f.
- Zugic, B.; Wang, L.; Heine, C.; Zakharov, D.N.; Lechner, B.A.J.; Stach, E.A.; Biener, J.; Salmeron, M.; Madix, R.J.; Friend, C.M. Dynamic restructuring drives catalytic activity on nanoporous gold–silver alloy catalysts. Nat. Mater. 2017, 16, 558-564, doi:10.1038/nmat4824.


