Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Efraín Santiago-Rodríguez | + 1184 word(s) | 1184 | 2021-12-28 04:16:44 | | | |
| 2 | Jessie Wu | Meta information modification | 1184 | 2021-12-28 07:16:37 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Santiago-Rodríguez, E. Patients with Juvenile Myoclonic Epilepsy. Encyclopedia. Available online: https://encyclopedia.pub/entry/17584 (accessed on 27 June 2026).
Santiago-Rodríguez E. Patients with Juvenile Myoclonic Epilepsy. Encyclopedia. Available at: https://encyclopedia.pub/entry/17584. Accessed June 27, 2026.
Santiago-Rodríguez, Efraín. "Patients with Juvenile Myoclonic Epilepsy" Encyclopedia, https://encyclopedia.pub/entry/17584 (accessed June 27, 2026).
Santiago-Rodríguez, E. (2021, December 28). Patients with Juvenile Myoclonic Epilepsy. In Encyclopedia. https://encyclopedia.pub/entry/17584
Santiago-Rodríguez, Efraín. "Patients with Juvenile Myoclonic Epilepsy." Encyclopedia. Web. 28 December, 2021.
Copy Citation
Juvenile myoclonic epilepsy (JME) appears in adolescence with myoclonic, absence, and generalized tonic clonic (GTC) seizures with paroxysmal activity of polyspike and slow wave (PSW), or spike and wave (SW) complexes in EEG.
juvenile myoclonic epilepsy
1. Background
Juvenile myoclonic epilepsy (JME) is a genetically-determined disease that appears in adolescence with myoclonic seizures, absence, and generalized tonic clonic (GTC) seizures that are characteristically present for two hours after awakening. Electroencephalogram (EEG) shows paroxysmal activity of polyspike and slow wave (PSW) or fast spike and wave complexes in frontocentral leads with a frequency of three- to five-hertz and a variable duration of events [1][2][3].
JME presents during childhood or adolescence with an absence of seizures in 40% of patients, and myoclonic and GTC seizures at some point in adolescence at approximately 17 years of age [4]. Some patients with JME have a family history of disease in variable ranges, depending on the population that is studied. Currently, some genes were postulated to partially explain the origin of the disease [5].
From a physiological point of view, it has been postulated that an imbalance exists between the excitatory and inhibitory neural networks that are located in the medial frontal regions, with a strong interrelation with the thalamocortical networks that regulate the sleep–wake cycle [6][7]. This alteration has been corroborated through EEG current source analysis studies, voxel-based morphometry, magnetic resonance spectroscopy, and functional magnetic resonance [8][9][10].
2. Clinical Characteristics
A total of 41 patients were selected, 16 men and 25 women, with a mean age of 21.1 ± 11.5 years (range of 12- to 68-years). A family history of epilepsy was present in 12 (29.2%) patients. The epilepsy age onset was of 13.6 ± 2.5 years. All the patients had myoclonic seizures with age onset at 13.6 ± 1.71 years. A total of 33 (80.4%) patients had GTC seizures with a mean onset age of 15.1 ± 0.8 years. Only nine (21.9%) patients had absence seizures with a mean age onset of 11.4 ± 1.5 years. Seizures are characteristically present in the first hours of the morning after awakening; our patients had seizures at 7:39 ± 3.2 a.m.
Patients with JME have many factors that increase the risk of seizures. In our group, the factors leading to seizures were sleep deprivation in 18 (43.9%) patients, alcoholic beverage intake in 8 (19.5%), stress in 7 (17.0%), photic stimulus in 1 (2.4%), menstruation in 1 (2.4%), and videogame playing in 1 (2.4%) patient. Only 4 patients (9.7%) had a diagnosis of JME at the time of entry into the study, and 23 (56.1%) patients had treatment with antiepileptic drugs. However, only 11 (26.8%) had treatment with valproate and 4 (9.7%) had treatment with levetiracetam, an antiepileptic drug that has demonstrated antiepileptic utility in JME. The mean daily dose of valproate was 661.7 ± 194.8 mg/day.
3. Paroxysmal Activity
All the patients had characteristic paroxysmal activity of the PSW complex; in addition, 30 (73.1%) patients had fast SW complexes, and 22 (53.6%) had slow waves. Although the duration of the EEG recordings was between 20- and 30-min, the number of events per hour was calculated. The mean number of paroxysmal events was 40.5/h ± 62.6 with a mean duration of 1.7 ± 1.1 s, the shortest duration of events was 0.3 s, and the longest duration was 15.2 s. The accumulated duration of paroxysmal events was 20.5 s, which represents 1.7% of all EEG recordings. The paroxysmal activity had asymmetrical distribution in 35 (85.4%) patients. Asymmetrical right paroxysmal activity was shown in 28 (68.3%) patients and 7 (17.0%) on the left side. Patients with asymmetric paroxysmal activity maintained such asymmetry throughout the EEG recording, but all patients had paroxysmal events on the opposite side. Focal paroxysmal events in temporal electrodes were observed in two patients (4.8%). None of the patients had myoclonic seizures during EEG recordings. The typical paroxysmal events are shown in Figure 1.
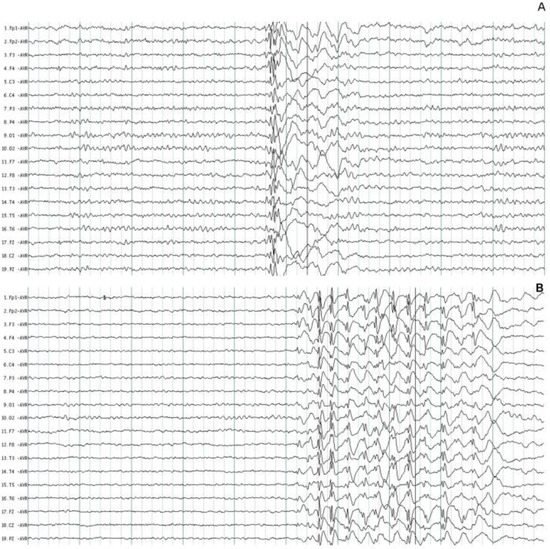
Figure 1. Typical paroxysmal activity in two patients with juvenile myoclonic epilepsy. (A) Polyspike and wave complexes of short duration. (B) Fast spike and wave complexes with more amplitude on the frontal leads. The normal background EEG activity was identified in two patients.
4. Background EEG Activity
The background EEG activity was normal in 28 (68.3%) patients (Figure 1) and abnormal only in 13 (31.7%) patients during visual EEG review. In these patients, the main abnormalities were generalized theta increases and decreases in alpha activity in six (46.1%) patients; the other six (46.1%) patients had generalized beta increases, and an increase in alpha activity was observed in only one (7.8%) patient. To determine if the use of antiepileptic treatment has any effect on the baseline EEG activity, a comparison was made between the proportion of patients with and without treatment. Of the 18 patients without antiepileptic treatment, 11 had a normal EEG; the 23 patients with antiepileptic treatment had 17 patients with a normal EEG. The differences were not significant (p = 0.38).
When quantitative EEG (QEEG) and neurometric analysis were used, more EEG abnormalities were found. Comparison of the proportion of patients with normal or abnormal EEG background activity between visual EEG analysis and QEEG showed a sensitivity of 30.8% and a specificity of 50.0%. Beta AP increases and delta AP decreases were the most frequent abnormalities that were found. In a broader description, increases were observed in beta AP in 22 (53.6%) patients, theta AP in 14 (34.1%), alpha AP in 13 (31.7%), and delta AP in 6 (14.6%). In contrast, a decrease in delta AP was found in 31 (75.6%) patients, theta AP in 23 (56.0%), beta AP in 18 (43.9%), and alpha AP in 15 (36.5%) patients (Figure 2).
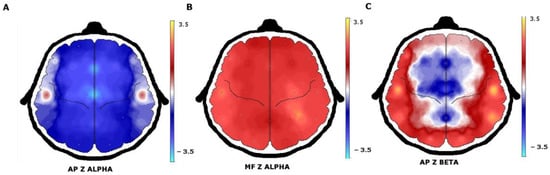
Figure 2. QEEG with neurometric analysis of patients with juvenile myoclonic epilepsy. (A) A generalized decrease in alpha absolute power in the frontocentral regions with (B) an increase in alpha mean frequency. (C) The beta absolute power increase was observed in the temporal and occipital regions.
When the analysis was carried out in patients without and with antiepileptic treatment, normal values were observed in the delta band in four patients without treatment and with antiepileptic treatment in two patients (p = 0.22). In the theta band, two patients without antiepileptic treatment had normal values, and three patients with treatment had normal values (p = 0.85). The beta band was normal in one patient in both groups (p = 0.85). Only in the alpha band were the differences significant, 2 patients without antiepileptic treatment had normal values and 11 patients with treatment (p = 0.012).
Abnormalities in MF were much less frequent than abnormalities in AP. An increase in theta MF was observed only in 11 (26.8%) patients, alpha MF in 8 (19.5%), and beta MF and delta MF in 6 (14.6%) patients. A decrease in delta MF was found in 18 (43.9%) patients, in alpha MF in 10 (24.1%), in theta MF in 6 (14.6%), and in beta MF in 5 (12.1%) patients. The number of patients who had normal MF values of the delta (p = 0.73), theta (p = 0.087), alpha (p = 0.48), and beta (p = 0.4) bands were not significantly different in those without and with antiepileptic treatment.
Spectral analysis showed an alpha peak in 9.91 ± 0.91 Hz with greater amplitude in the occipital regions on the O1 and O2 electrodes. Almost half of the patients (48.7%) had normal results when they were compared with the spectral alpha peak in 9.766 or 10.156 Hz. Only 11 (26.8%) patients had shifts at higher frequencies above 10.156 Hz and 10 (24.4%) below 9.766 Hz. A typical shift to lower frequencies of the alpha spectral peak is shown in Figure 3. The alpha spectral peak had normal values in 8 patients without antiepileptic treatment and in 12 patients with treatment, these differences were not significant (p = 0.62).
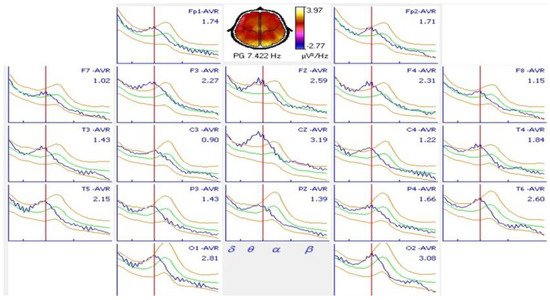
Figure 3. Spectral analysis of an 18-year-old patient with juvenile myoclonic epilepsy and comparison with normal values. A shift in the alpha spectral peak to a 7.422 Hz frequency was observed in all derivations. A normal alpha spectral peak at this age occurs at 10.156 Hz.
References
- Janz, D. Epilepsy with impulsive petit mal (juvenile myoclonic epilepsy). Acta Neurol. Scand. 1985, 72, 449–459.
- Delgado-Escueta, A.V.; Enrile-Bacsal, F. Juvenile myoclonic epilepsy of Janz. Neurology 1984, 34, 285–294.
- Trenité, D.G.K.-N.; Schmitz, B.; Janz, D.; Delgado-Escueta, A.V.; Thomas, P.; Hirsch, E.; Lerche, H.; Camfield, C.; Baykan, B.; Feucht, M.; et al. Consensus on diagnosis and management of JME: From founder’s observations to current trends. Epilepsy Behav. 2013, 28, S87–S90.
- Genton, P.; Thomas, P.; Kasteleijn-Nolst Trenité, D.G.; Medina, M.T.; Salas-Puig, J. Clinical aspects of juvenile myoclonic epilepsy. Epilepsy Behav. 2013, 28, S8–S14.
- Santos, B.; Marinho, C.R.M.; Marques, T.E.B.S.; Angelo, L.K.G.; Malta, M.V.D.S.; Duzzioni, M.; De Castro, O.W.; Leite, J.P.; Barbosa, F.T.; Gitaí, D.L.G. Genetic susceptibility in Juvenile Myoclonic Epilepsy: Systematic review of genetic association studies. PLoS ONE 2017, 21, 12–18.
- Wolf, P.; Yacubian, E.M.T.; Avanzini, G.; Sander, T.; Schmitz, B.; Wandschneider, B.; Koepp, M. Juvenile myoclonic epilepsy: A system disorder of the brain. Epilepsy Res. 2015, 114, 2–12.
- Yacubian, E.M. Juvenile myoclonic epilepsy: Challenges on its 60th anniversary. Seizure 2017, 44, 48–52.
- Anderson, J.; Hamandi, K. Understanding juvenile myoclonic epilepsy: Contributions from neuroimaging. Epilepsy Res. 2011, 94, 127–137.
- Wandschneider, B.; Thompson, P.J.; Vollmar, C.; Koepp, M.J. Frontal lobe function and structure in juvenile myoclonic epilepsy: A comprehensive review of neuropsychological and imaging data. Epilepsia 2012, 53, 2091–2098.
- Santiago-Rodrıguez, E.; Harmony, T.; Fernández-Bouzas, A.; Hernández-Balderas, A.; Martınez-López, M.; Graef, A.; Garcıa, J.C.; Fernández, T. Source analysis of polyspike and wave complexes in juvenile myoclonic epilepsy. Seizure 2002, 11, 320–324.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
920
Revisions:
2 times
(View History)
Update Date:
28 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No