Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yuliya Leonardovna Vechtomova | + 2035 word(s) | 2035 | 2021-11-02 09:11:54 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Vechtomova, Y. UV Radiation in DNA Damage and Repair. Encyclopedia. Available online: https://encyclopedia.pub/entry/17565 (accessed on 08 August 2026).
Vechtomova Y. UV Radiation in DNA Damage and Repair. Encyclopedia. Available at: https://encyclopedia.pub/entry/17565. Accessed August 08, 2026.
Vechtomova, Yuliya. "UV Radiation in DNA Damage and Repair" Encyclopedia, https://encyclopedia.pub/entry/17565 (accessed August 08, 2026).
Vechtomova, Y. (2021, December 27). UV Radiation in DNA Damage and Repair. In Encyclopedia. https://encyclopedia.pub/entry/17565
Vechtomova, Yuliya. "UV Radiation in DNA Damage and Repair." Encyclopedia. Web. 27 December, 2021.
Copy Citation
Prolonged exposure to ultraviolet radiation on human skin can lead to mutations in DNA, photoaging, suppression of the immune system, and other damage up to skin cancer (melanoma, basal cell, and squamous cell carcinoma).
DNA repair
cancer
ultraviolet
ROS
1. Introduction
The constant destructive impact of various adverse environmental factors (pollution with toxic substances, various types of natural and artificial radiation, etc.) causes the disturbance of the normal functioning of living cells [1][2][3]. This leads to the development of various diseases, which can eventually result in chronic ones. In turn, the immune and endocrine systems cease to cope with their protective and regulatory functions due to an increase in constant load, and, ultimately, all this can lead to more serious disorders, including cancer. A necessary condition for the occurrence and development of the process of carcinogenesis is DNA mutations. DNA mutations can occur due to mutagenic environmental factors (in particular, UV) when the effective repair does not function [4][5][6]. A person is exposed to intense UV light both in connection with professional activities that require a long stay out of doors and as a result of following fashion trends, sunbathing on the beach, or using special lamps in tanning salons. Prolonged exposure to UV on the skin leads to hyperpigmentation, photoaging due to collagen fiber damage [7], and the accumulation of mutations in the cell’s DNA. About 90% of non-melanoma skin cancers and 86% of melanomas are associated with chronic UV irradiation of the skin [8]. UV inhibits the synthesis of ATP and disrupts the immune response, which also contributes to carcinogenesis [8][9]. It was found that chronic exposure to solar radiation is the most important environmental factor involved in the pathogenesis of actinic keratosis and squamous cell carcinoma [9]. It has also been shown that chronic UV radiation in farmers is associated with a high risk of the earlier development of basal cell carcinoma and its aggressive subtypes [10].
2. Possible Mechanisms of DNA Damage by UV Radiation
2.1. Target Molecules for UV Exposure
Targets of UV radiation in living organisms can be various photoactive molecules, for example, pterins, folates, flavins, porphyrins, aromatic amino acids, etc. (Figure 1), as well as biopolymers: proteins and nucleic acids [11][12]. Such photoactive molecules transfer into an excited state after the light absorption, and the excess energy can be utilized in several ways, among which three main options are of biological significance.
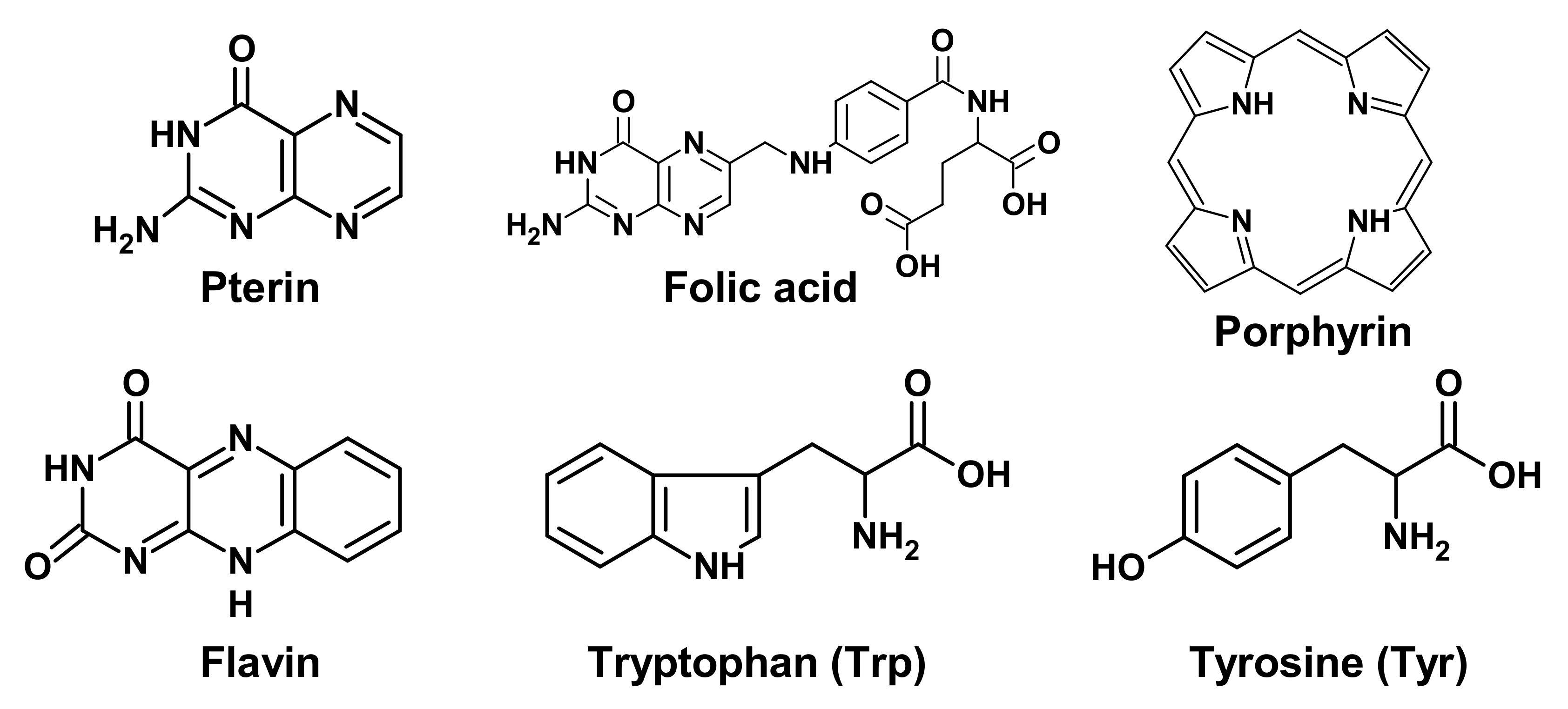
Figure 1. Chemical formulas of low molecular weight biological chromophores.
(1) If the molecule is a chromophore of a photoreceptor protein, then the absorbed energy is converted into a signal to trigger various processes. For example, flavin is a chromophore of cryptochromes (CRYs), which are involved in the photoregulation of circadian rhythms.
(2) Important biologically active molecules can undergo partial chemical modification or be completely destroyed, which can lead to a deficiency of these molecules in the body. This is especially true for those substances that cannot be synthesized in the human body. For example, folic acid derivatives are destroyed by UV radiation. This may be the reason for the deficiency of this vitamin in people with fair skin, exposed to increased impact to solar radiation [13].
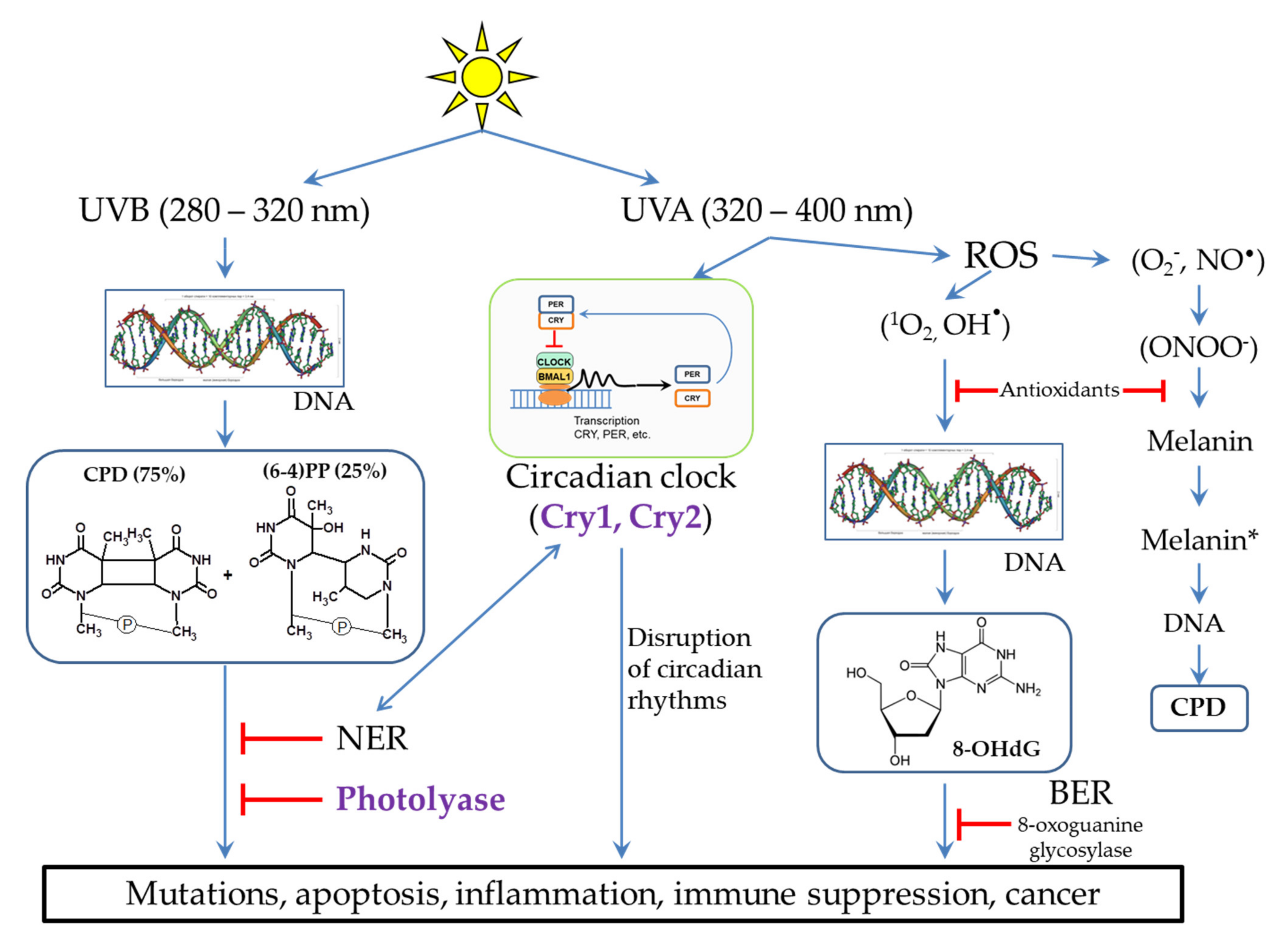
Figure 2. Different UV radiation of DNA leads to different lesions and subsequent occurrence of pathological situations. Methods of lesion prevention and repair are presented. * - The excited state of melanin.
(3) Light-excited molecules can become sensitizers for the destruction of other molecules or lead to the formation of reactive oxygen species (ROS). ROS, in turn, can lead to damage to other molecules, including DNA (Figure 2) and lipids [14]. Porphyrins (Figure 1) are well-known photosensitizers in medicine, and their ability to generate ROS underlies the photodynamic cancer therapy [15].
2.2. Damaging Effect of UVB Light on DNA
The most dangerous impact of UVB (280–320 nm) radiation is the DNA damage in skin cells. The main products of UVB-irradiated DNA are cyclobutane pyrimidine dimers (CPD, 75%) and pyrimidine-pyrimidone (6-4) photoproducts ((6-4)PP, 25%), in which two neighboring pyrimidines are covalently bound (Figure 2). In addition, irradiation can lead to breaks in one or both of DNA chains at once. The formation and accumulation of CPD and (6-4) PP block the DNA replication and transcription, which disrupts the normal functioning of cells. If these lesions are not removed in a timely manner, this can lead to cell death and the occurrence of inflammatory processes at the tissue level. In some cases, errors in the repair process can lead to the appearance and accumulation of mutations (see Section 3), which can subsequently cause various diseases and skin cancer [16][17][18][19][20].
2.3. The Influence of UVA Light on the Processes Occurring in Skin Cells
It is believed that UVA radiation (320–400 nm) is not as harmful as UVB. In small doses, UVA light is necessary for a human because it is a signal for various photoregulatory proteins, in particular for restarting circadian rhythms, which, in turn, regulate many different processes in the body (Figure 2). However, in high doses, UVA radiation can lead to the suppression of the immune system and the formation of ROS through photosensitization reactions [9][14][18][21]. ROS cause oxidative stress and photoaging, and this can also lead to skin cancer [20]. DNA can be damaged by ROS to form 8-oxo-7,8-dihydro-2’-deoxyguanosine (8-OHdG) (Figure 2), which, such as CPD, interferes with normal cell functioning [9][14][18][20][21]. ROS activate the expression of matrix metalloproteinases, which cause degradation of collagen fibers, leading to the appearance of wrinkles and skin aging [7][20][22][23].
A special pathway of the destructive effect of UVA radiation on DNA is caused by cell damage in the presence of melanin (Figure 2). Back in 2003, it was shown that oxidative damage is not the main type of UVA-induced damage in skin cancer. Thus, oxidized pyrimidines, single-chain breaks, oxidized purines (8-OHdG), and CPD are formed in a ratio of 1:1:3:10. Moreover, it was found that UVA generates CPD with a large predominance of thymine dimers, which indicates their formation through photosensitized triplet energy transfer [24]. In further studies, it was shown that melanin is the photosensitizer of the process. Melanin properties are two-fold: 1) when the melanin synthesis is completed, melanin in a certain geometric configuration in keratinocytes performs a protective function, but 2) it can have pro-oxidant properties during partial polymerization in melanocytes when exposed to UV radiation. The potential role of UVA in skin carcinogenesis is also confirmed by epidemiological studies showing an increased risk of melanoma among the users of tanning lamps producing UVA radiation [25].
In pigmented melanocytes, CPD occurs both instantly and within a few hours after UV irradiation in the dark. The path of CPD occurrence in the dark is partially similar to bioluminescence and is as follows. UV activates nitric oxide synthases, which generate a nitric oxide radical (NO•), and NADPH oxidases, which generate superoxide-anion radical (O2−). Further, these radicals interact with each other to form strong oxidant peroxynitrite (ONOO−) (Figure 2). Peroxynitrite oxidizes melanin while exciting the melanin electron to a high-energy level. This is the process of “chemical electron excitation” in melanin: a non-radiative triplet energy transfer to DNA occurs with the formation of CPD [26][27]. UVA and peroxynitrite contribute to the solubilization of melanin and increase the permeability of the nuclear membrane to melanin. This pathway can be considered as a melanin-dependent pathogenesis of melanoma. In the same way, the chemical excitation of melanin can trigger pathogenesis in other tissues, in which nitric oxide and superoxide anion radicals arise in cells containing melanin [28]. It is important to note that photodynamic therapy of skin cancer with red light does not cause CPD formation in the presence of melanin [29].
Exposure to strong UV radiation is the main etiological environmental factor for all forms of skin cancer, including melanoma. The ability to repair DNA determines the risk of skin cancer. The sensitivity of cells to the severe effects of UV radiation depends on the degree of skin pigmentation. In turn, the process of melanogenesis can be disrupted when exposed to various exogenous etiological environmental factors, including UV [30]. Disruption of melanogenesis occurs in a number of dermatological diseases, including vitiligo [31][32][33]. The study of melanogenesis and the ways of its regulation are important for the development of new photoprotective strategies for the prevention of skin cancer.
3. DNA Repair Systems Involved in the Photodamage Removal
Cells have many different mechanisms for repairing each type of DNA damage that occurs spontaneously and is caused by exogenous factors [6][34]. The main mechanisms of DNA repair include: direct repair, when the enzyme restores the original structure without removing damaged nucleotides; excision repair, through the removal of damaged sites, followed by the synthesis of new nucleotides: base excision repair (BER) and nucleotide excision repair (NER); repair of unpaired bases (mismatch repair); repair of single-strand and double-strand breaks [6][34]. To remove photodamage, both direct repair with the participation of the DNA photolyase [4][35][36] and NER for CPD and (6-4)PP or BER for 8-OHdG [4][20][37] are used (Figure 3).
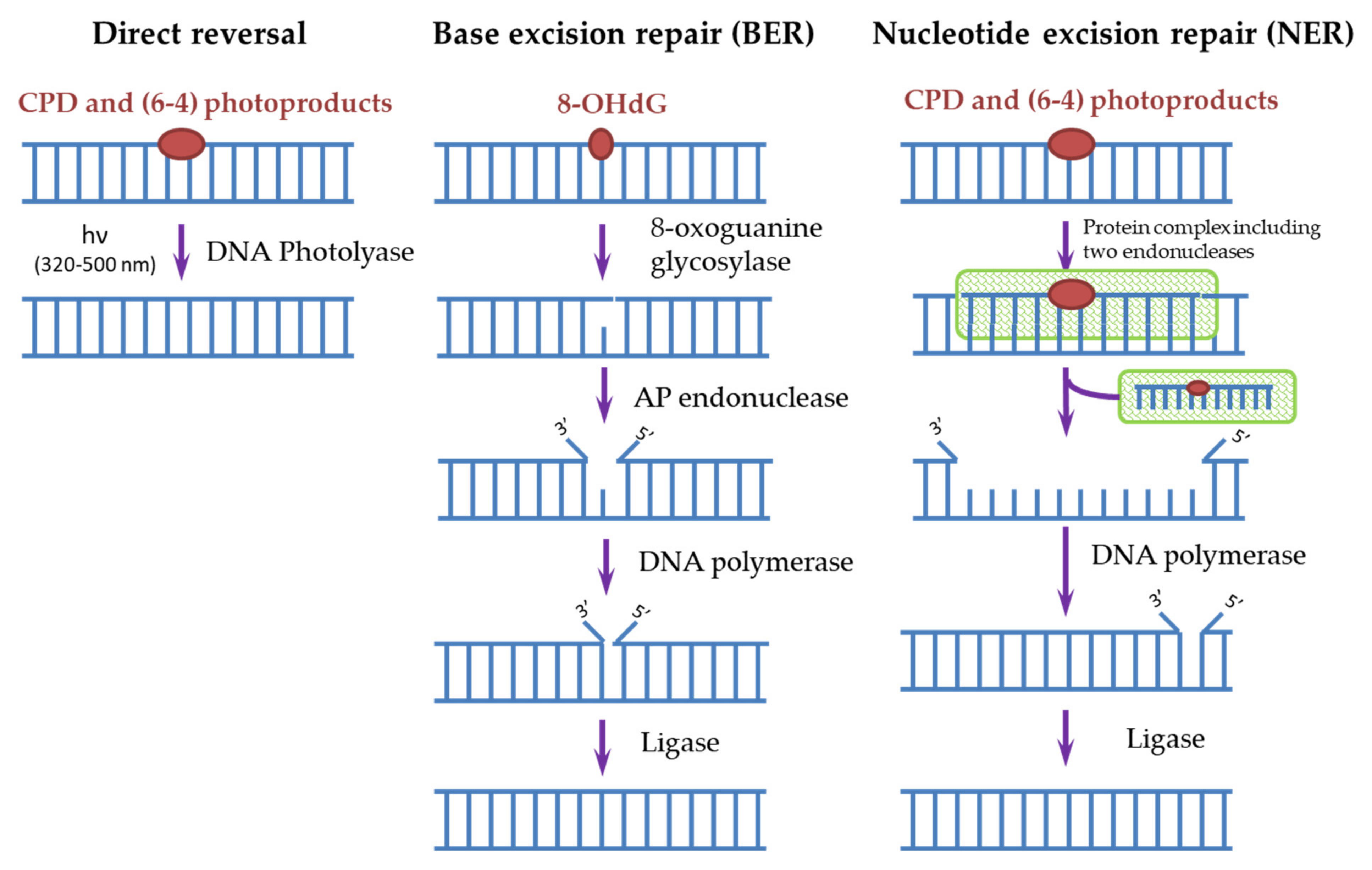
Figure 3. Scheme of direct and excisional repair in photodamaged DNA.
In humans, as in most mammals (except for some marsupials), there are no DNA photolyases and the only system responsible for removing the most dangerous CPDs for DNA is NER. It is known that defects in the NER process lead to a xeroderma pigmentosum disease (XP). People suffering from this disease are extremely sensitive to sunlight and are susceptible to the development of skin oncological diseases. Genetic analysis of such patients revealed the mutations in seven main genes, called XPA-XPG, responsible for the NER function [35]. In general, more than 20 different proteins take part in the NER process, and it can be triggered in two ways. The first way is global genome NER when the NER enzymes (in humans, these are complexes of RPA, XPA, and XPC proteins) themselves find damaged sites and start the repair process. The second way is transcription-coupled NER, where, during transcription, an RNA polymerase bumps into a damaged site and initiates the repair process. Next, a cascade of reactions involving a protein complex is started, as a result of which endonucleases (XPF and XPG) cut out a DNA oligomer of about 30 nucleotides containing CPD. Then, a polymerase is attached and synthesizes a complementary intact DNA chain. The ligase completes the process by connecting the free ends of DNA chains (Figure 3) [4][35][37][38][39]. Both pathways require the involvement of a large number of enzymes, and as a result, the process stretches over time to several hours, thus the processes triggered by CPD dimers, for example, melanogenesis, have time to start in cells. In addition, the repair process itself often occurs with violations and leads to mutations. In such erroneous CPD repair, mutations with cytosine-to-thymine replacement occur most often, which is characteristic for mutations found in cancer cells [9][17][40][41].
The BER system is used to remove 8-OHdG formed as a result of photo-oxidative stress (Figure 3). The BER scheme is similar to the NER scheme but includes fewer enzymes. The damaged base is removed by 8-oxoguanine glycosylase, then from 1 to 13 nucleotides near the damaged site are removed by AP endonuclease and synthesized again. Disruption in the BER function leads to fetal mortality or predisposition to cancer and neurological symptoms in animals. NER is able to partially replace BER in case of violations in its operation and also stimulate the enzymatic activity of some BER factors [37].
Furthermore, sometimes the NER system cannot recognize CPD and restore DNA, in this case, a rough “SOS” repair system works: during replication, the affected DNA sections are bypassed and subsequently replaced with nucleotides that are not complementary to the original chain, which can also lead to mutations [42]. Most of the spontaneously occurring mutations that accumulate in cells throughout a person’s life can go unnoticed without any serious consequences, but some of them can change key cellular functions and lead to cancer and aging [9].
Understanding the function of repair systems is important not only because of possible disruptions in their work that lead to the occurrence of oncological diseases but also because of possible mechanisms and places of application in the treatment of these diseases. Many cancer treatment strategies are aimed to destroy the DNA of tumor cells, and in this case, the repair systems existing in these cells will reduce the effectiveness of such treatment [6]. At the same time, during chemotherapy and radiotherapy, not only diseased cells are often affected, but also healthy ones. The ability to speed up the process of restoring these cells after the end of treatment (during the rehabilitation period) will also improve the quality of treatment.
References
- Anwar, F.; Chaudhry, F.; Nazeer, S.; Zaman, N.; Azam, S. Causes of Ozone Layer Depletion and Its Effects on Human: Review. Atmos. Clim. Sci. 2016, 6, 129–134.
- Marrot, L. Pollution and Sun Exposure: A Deleterious Synergy. Mechanisms and Opportunities for Skin Protection. Curr. Med. Chem. 2018, 25, 5469–5486.
- Araviiskaia, E.; Berardesca, E.; Bieber, T.; Gontijo, G.; Sanchez Viera, M.; Marrot, L.; Chuberre, B.; Dreno, B. The impact of airborne pollution on skin. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1496–1505.
- Morita, R.; Nakane, S.; Shimada, A.; Inoue, M.; Iino, H.; Wakamatsu, T.; Fukui, K.; Nakagawa, N.; Masui, R.; Kuramitsu, S. Molecular mechanisms of the whole DNA repair system: A comparison of bacterial and eukaryotic systems. J. Nucleic Acids. 2010, 2010, 179594.
- Cadet, J.; Douki, T. Formation of UV-induced DNA damage contributing to skin cancer development. Photochem. Photobiol. Sci. 2018, 17, 1816–1841.
- Huang, R.; Zhou, P.-K. DNA damage repair: Historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 254.
- Kuznetsova, E.V.; Snarskaya, E.S.; Zavalishina, L.E.; Tkachenko, S.B. Immunohistochemical study of the specific features of expression of matrix metalloproteinases 1, 9 in the photoaged skin, the foci of actinic keratosis and basal cell carcinoma. Arkh. Patol. 2016, 78, 17–22.
- Yeager, D.G.; Lim, H.W. What’s New in Photoprotection: A Review of New Concepts and Controversies. Dermatol. Clin. 2019, 37, 149–157.
- Leccia, M.T.; Lebbe, C.; Claudel, J.P.; Narda, M.; Basset-Seguin, N. New Vision in Photoprotection and Photorepair. Dermatol. Ther. (Heidelb) 2019, 9, 103–115.
- Apalla, Z.; Lallas, A.; Sotiriou, E.; Lazaridou, E.; Vakirlis, E.; Trakatelli, M.; Kyrgidis, A.; Ioannides, D. Farmers develop more aggressive histologic subtypes of basal cell carcinoma. Experience from a Tertiary Hospital in Northern Greece. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 17–20.
- Buglak, A.A.; Telegina, T.A.; Kritsky, M.S. A quantitative structure-property relationship (QSPR) study of singlet oxygen generation by pteridines. Photochem. Photobiol. Sci. 2016, 15, 801–811.
- Fu, S.; Xue, S.; Chen, J.; Shang, S.; Xiao, H.; Zang, Y.; Tang, X. Effects of Different Short-Term UV-B Radiation Intensities on Metabolic Characteristics of Porphyra haitanensis. Int. J. Mol. Sci. 2021, 22, 2180.
- Jablonski, N.G.; Chaplin, G. Colloquium paper: Human skin pigmentation as an adaptation to UV radiation. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. S2), 8962–8968.
- Di Mascio, P.; Martinez, G.R.; Miyamoto, S.; Ronsein, G.E.; Medeiros, M.H.G.; Cadet, J. Singlet Molecular Oxygen Reactions with Nucleic Acids, Lipids, and Proteins. Chem. Rev. 2019, 119, 2043–2086.
- Kwiatkowski, S.; Knap, B.; Przystupski, D.; Saczko, J.; Kędzierska, E.; Knap-Czop, K.; Kotlińska, J.; Michel, O.; Kotowski, K.; Kulbacka, J. Photodynamic therapy—Mechanisms, photosensitizers and combinations. Biomed. Pharmacother. 2018, 106, 1098–1107.
- Stege, H.; Roza, L.; Vink, A.A.; Grewe, M.; Ruzicka, T.; Grether-Beck, S.; Krutmann, J. Enzyme plus light therapy to repair DNA damage in ultraviolet-B-irradiated human skin. Proc. Natl. Acad. Sci. USA 2000, 97, 1790–1795.
- Harrison, C.B.; O’Neil, L.L.; Wiest, O. Computational studies of DNA photolyase. J. Phys. Chem. A 2005, 109, 7001–7012.
- Khan, A.Q.; Travers, J.B.; Kemp, M.G. Roles of UVA radiation and DNA damage responses in melanoma pathogenesis. Environ. Mol. Mutagen. 2018, 59, 438–460.
- Johann To Berens, P.; Molinier, J. Formation and Recognition of UV-Induced DNA Damage within Genome Complexity. Int. J. Mol. Sci. 2020, 21, 6689.
- Luze, H.; Nischwitz, S.P.; Zalaudek, I.; Müllegger, R.; Kamolz, L.P. DNA repair enzymes in sunscreens and their impact on photoageing—A systematic review. Photodermatol. Photoimmunol. Photomed. 2020, 36, 424–432.
- Cadet, J.; Douki, T.; Ravanat, J.L. Oxidatively generated damage to cellular DNA by UVB and UVA radiation. Photochem. Photobiol. 2015, 91, 140–155.
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868.
- Freitas-Rodríguez, S.; Folgueras, A.R.; López-Otín, C. The role of matrix metalloproteinases in aging: Tissue remodeling and beyond. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 2015–2025.
- Douki, T.; Reynaud-Angelin, A.; Cadet, J.; Sage, E. Bipyrimidine photoproducts rather than oxidative lesions are the main type of DNA damage involved in the genotoxic effect of solar UVA radiation. Biochemistry 2003, 42, 9221–9226.
- Beani, J.C. Ultraviolets A et dommages de l’ADN; leur place dans la cancérogenèse cutanée . Bull. Acad. Natl. Med. 2014, 198, 273–295.
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.; Halaban, R.; Douki, T.; Brash, D.E. Photochemistry. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847.
- Premi, S.; Brash, D.E. Chemical excitation of electrons: A dark path to melanoma. DNA Repair 2016, 44, 169–177.
- Brash, D.E. UV-induced Melanin Chemiexcitation: A New Mode of Melanoma Pathogenesis. Toxicol. Pathol. 2016, 44, 552–554.
- Mudambi, S.; Pera, P.; Washington, D.; Remenyik, E.; Fidrus, E.; Shafirstein, G.; Bellnier, D.; Paragh, G. Photodynamic therapy does not induce cyclobutane pyrimidine dimers in the presence of melanin. Photodiagnosis Photodyn. Ther. 2018, 22, 241–244.
- Swope, V.B.; Abdel-Malek, Z.A. MC1R: Front and Center in the Bright Side of Dark Eumelanin and DNA Repair. Int. J. Mol. Sci. 2018, 19, 2667.
- Bidaki, R.; Majidi, N.; Moghadam Ahmadi, A.; Bakhshi, H.; Sadr Mohammadi, R.; Mostafavi, S.A.; Kazemi Arababadi, M.; Hadavi, M.; Mirzaei, A. Vitiligo and social acceptance. Clin. Cosmet. Investig. Dermatol. 2018, 11, 383–386.
- Telegina, T.A.; Lyudnikova, T.A.; Buglak, A.A.; Vechtomova, Y.L.; Biryukov, M.V.; Demin, V.V.; Kritsky, M.S. Transformation of 6-tetrahydrobiopterin in aqueous solutions under UV-irradiation. J. Photochem. Photobiol. A. 2018, 354, 155–162.
- Telegina, T.A.; Vechtomova, Y.L.; Kritsky, M.S.; Madirov, E.I.; Nizamutdinov, A.S.; Obuhov, Y.N.; Buglak, A.A. Tetrahydrobiopterin Photooxidation: A Key Process in Vitiligo Phototherapy. Appl. Biochem. Microbiol. 2021, 57, 571–578.
- Prorok, P.; Grin, I.R.; Matkarimov, B.T.; Ishchenko, A.A.; Laval, J.; Zharkov, D.O.; Saparbaev, M. Evolutionary Origins of DNA Repair Pathways: Role of Oxygen Catastrophe in the Emergence of DNA Glycosylases. Cells 2021, 10, 1591.
- Sancar, A. Mechanisms of DNA Repair by Photolyase and Excision Nuclease (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 2016, 55, 8502–8527.
- Kavakli, I.H.; Ozturk, N.; Gul, S. DNA repair by photolyases. Adv. Protein Chem. Struct. Biol. 2019, 115, 1–19.
- Lee, T.H.; Kang, T.H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092.
- Jans, J.; Schul, W.; Sert, Y.G.; Rijksen, Y.; Rebel, H.; Eker, A.P.; Nakajima, S.; van Steeg, H.; de Gruijl, F.R.; Yasui, A.; et al. Powerful skin cancer protection by a CPD-photolyase transgene. Curr. Biol. 2005, 15, 105–115.
- Guintini, L.; Charton, R.; Peyresaubes, F.; Thoma, F.; Conconi, A. Nucleosome positioning, nucleotide excision repair and photoreactivation in Saccharomyces cerevisiae. DNA Repair 2015, 36, 98–104.
- Taylor, J.-S. Unraveling the Molecular Pathway from Sunlight to Skin Cancer. Acc. Chem. Res. 1994, 27, 76–82.
- Boros, G.; Miko, E.; Muramatsu, H.; Weissman, D.; Emri, E.; Rózsa, D.; Nagy, G.; Juhász, A.; Juhász, I.; van der Horst, G.; et al. Transfection of pseudouridine-modified mRNA encoding CPD-photolyase leads to repair of DNA damage in human keratinocytes: A new approach with future therapeutic potential. J. Photochem. Photobiol. B 2013, 129, 93–99.
- Maslowska, K.H.; Makiela-Dzbenska, K.; Fijalkowska, I.J. The SOS system: A complex and tightly regulated response to DNA damage. Environ. Mol. Mutagen. 2019, 60, 368–384.
More
Information
Subjects:
Dermatology; Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
4.6K
Revision:
1 time
(View History)
Update Date:
27 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No