 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rubén Rabaneda Bueno | + 4468 word(s) | 4468 | 2021-12-13 04:05:41 |
Video Upload Options
Alzheimer's disease (AD) is an adult-onset dementia characterised by progressive neurodegeneration and widespread brain damage, leading to long-term functional and cognitive impairment and greatly reduced life expectancy. While early genetic studies uncovered several polymorphisms associated with AD, more recently genome-wide association analyses and massive sequencing techniques have revealed numerous novel susceptibility genes, differentially expressed genes, and disease traits. Nevertheless, the mechanisms underlying disease onset are still not fully understood and, as with other complex human diseases, the causes of low heritability are unclear. Epigenetic mechanisms, in which changes in gene expression do not depend on changes in genotype, have been postulated as key factors in understanding the development of AD and the processes that influence age-related changes and various neurological diseases. Research on specific mutations in risk genes and epigenetic markers is increasing, enabling the development of therapeutic treatments that target the neuropathological changes associated with AD and, in many cases, are expected to reverse at least some of the cognitive impairment associated with the disease. The application of effective therapies therefore requires a growing understanding of the genetic risk factors and underlying epigenetic mechanisms involved in AD.
1. Alzheimer's Disease and the Genome: Epistasis and Key Susceptibility Genes-Targeted Therapies.
Alzheimer's disease (AD) is characterized by the accumulation of Aβ-peptide and the formation of NFTs, preceded by the expression of a number of genetic factors that interact through complex biochemical pathways [1]. The rare familial form of AD occurs in less than 6% of patients, of whom approximately 60% have a family history of AD and 13% have an autosomal dominant inheritance. This form of AD occurs at less than 65 years of age as Early Onset AD (EOAD), usually in the thirties or forties. The prevalence of neurodegenerative dementia associated with EOAD is characterized by rapid disease onset and shorter estimated survival [2], which worsens as patients age and reach the age group of 65 years [3]. This has been associated with the deposition of phosphorylated tau protein (p-Tau) in the brain at a very early stage [4]. In contrast, in the multifactorial sporadic form, susceptibility is determined by multiple genetic factors interacting with environmental factors. This form usually occurs as a late onset AD (LOAD) around the age of 65 and is genetically more complex, both in terms of inheritance and etiopathology [5][6][7].
1.1. Major Susceptibility Genes
Candidate gene studies have compared the frequency of genetic variants and identified the protein-coding genes of the amyloid-β precursor protein (APP) and the PS1 and PS2 subunits of presenilins (PSEN1, PSEN2) and apolipoprotein E (ApoE) [8][9][10] as important susceptibility factors for AD. The most common allelic form of ApoE is ε3 (ApoE3), followed by the ε4 allele (ApoE4), which is the predominant risk factor for sporadic AD, and the ε2 allele (ApoE2) is the rarest form. In the European population, the observed allele frequencies are 74.98% ApoE3, 8.62% ApoE2 and 16.40% ApoE4 in individuals under or equal to 65 years of age and 78.74%, 7.74% and 13.52%, respectively, in individuals above this age [11]. Substitution of several amino acids leads to a structural change that promotes binding to very low density lipoproteins (VLDL) in ApoE4 and to high density lipoproteins (HDL) in ApoE3 and ApoE2. In the case of ApoE2, this structural affinity for HDL determines its neuroprotective properties, especially in women, who have higher levels of HDL [12], insulin-like growth factor I (IGF-I) and glucose transporter type 4 (GLUT4) compared to the other two isoforms [13]. The genetic risk of ApoE is also age-dependent, with the ApoE4/ApoE4 genotype generally occurring in individuals aged 65 years and the ApoE3/ApoE4 genotype occurring in individuals aged 85 years, with no significant differences between the sexes [10][14]. ApoE4 has an estimated heritability of 0.13-0.33 and about 25% of overall heritability (LOAD), but these estimates tend to increase significantly in twin studies [15][16][17].Autosomal dominant variants of PSEN1 and PSEN2 genes and Aβ precursor genes are responsible for 5-10% of EOAD cases, while the remaining cases of AD are explained by the effects of different polygenic variants in an additive model [18]. These genes are expressed through common biological pathways with Aβ metabolism, so mutations in them have great potential to influence amyloid pathogenicity and early onset [19]. A missense variant of APP at codon 673 increases the risk of AD [20], and another in PSEN1 alters the processing of APP and promotes the accumulation of Aβ plaques [17]. Mutations in the Aβ sequence of APP can promote fibrillation and early cognitive impairment or cause inhibition of Aβ-peptide that prevents neuronal dysfunction [21].
Bridging integrator 1 (BIN1) is the second major risk locus of AD, and increased expression has been associated with cognitive impairment and accelerated disease onset due to tauopathy [22][23][24]. CLU is considered the third most important genome-wide risk locus for LOAD, which influences the disease progression of MCI [25]. Along with variants of CD33, MS4A4, and CD2AP [26], they have been found to increase the risk of LOAD in Caucasian populations but not in Asian populations, or association studies in this population have been inconsistent. CLU is particularly regulated by interactions with other risk loci involved in various regulatory processes of tau pathology, such as Aβ clearance, Aβ binding and deposition [27][28] and cerebral neuroinflammatory stress responses [29][30][31]. Alone or in interaction with other loci, increased expression of CLU leads to neuronal dysfunction and amyloid plaque formation [32][33][34]. There are other important protein-coding genes that increase the risk of LOAD, such as PICALM, APOC1, SORL1, CR1, ABCA7 and ESR1 [17][35][36][37][38][39], as well as differentially expressed genes in brain regions associated with amyloid pathologies, such as the gene SERPINA3. In particular, this gene encodes the α1-antichymotrypsin protein, which is upregulated in the brain of AD patients [40][41] and exerts an inhibitory effect on serine protease enzymes associated with dementia risk [42]. SERPINA3 induces neuronal death by promoting increased p-Tau levels, Aβ deposition and the formation of NFTs [43][44][45].
NGS sequencing studies documented several associations between TREM2 and EOAD and LOAD forms in Caucasian populations, with a number of variants increasing the risk of LOAD two- to fourfold, comparable to that of an ApoE4 carrier [46][47]. R47H is the best known variant of TREM2, which significantly increases the risk of LOAD and promotes the rapid onset of symptoms and cognitive impairment. The effects of this variant appear to be associated with amyloid pathology and NFTS formation, particularly in Caucasian populations. Increased levels of total tau and p-Tau have been found in the cerebrospinal fluid (CSF) of R47H carriers compared to non-carriers [48].
There are mutations in other genes such as nonsense variants in the LOAD susceptibility genes PSEN1, PSEN2, MAPT, PRNP, CSF1R, and GRN that have been associated with various neurodegenerative diseases in Asian populations. These mutations associated with neurodegenerative diseases should also be investigated in addition to the risk loci in the clinical diagnosis of [49]. Variants of the gene CD2AP have been associated with EOAD and are also thought to play a critical role in the development of AD [50][51]. Recent studies on gene haplotypes have shown a strong influence of the regulated expression of the MAPT gene in the brain [52] and differences between haplotypes of TOMM40 encoding ApoE4 and ApoE3 on the association probability for AD [53].
1.2. Epistatic interactions between Risk Loci
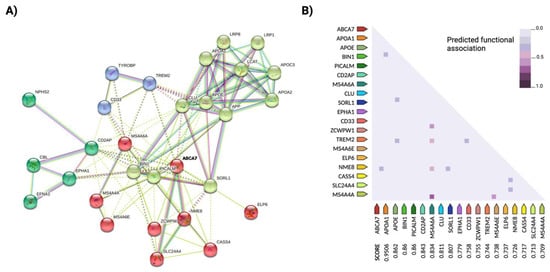
As with many complex human traits, only a minimal portion of the heritability of AD [54] appears to be explained by genetic risk factors. One explanation for this "missing heritability" could be the occurrence of rare and undetected variants such as mutations in ADAM10 [55], which contribute greatly to the amplification of the disease phenotype. As in other complex diseases, susceptibility to AD is determined by additive effects and epistatic interactions of multiple genetic variants. It has also been proposed that TREM2 and ApoE jointly influence the pathogenesis of LOAD, although the mechanisms of interaction between the two risk loci are still unknown [29][56] BIN1 interacts with coding products of similar functional pathways affected in AD, such as PICALM, MAPT, ABCA7 or SORL1; CLU and ApoE have additive effects on lipid trafficking and affect Aβ deposition, and ABCA7 interacts with the major risk candidate genes BIN1, CLU and PICALM [6] (Figure 1). Genetic interactions between the rs670139 and rs11136000 polymorphisms of the MS4A4E and CLU genes predict up to 8% increased risk for the occurrence of AD [57]. Indeed, the epistatic dominance effect of MS4A4E-CLU, together with ApoE, APP and TREM2, is among the most important risk loci identified to date.
Updated multivariate GWAS (MGAS) opens new possibilities in the study of complex traits and can contribute to the discovery of new susceptibility genes and to a better understanding of the interactions between known risk genes. This type of analysis uses information on biological relationships in combination with bioinformatics tools such as a network analysis of protein interactions (PPI) [58][59] (see Figure 1 for an example of a PPI network analysis in AD). For example, the risk genes ApoE and APOC1 were found to be related to eight subcortical measures of the AD neuroimaging phenotype after applying a multivariate screen based on the extended Simes method (GATES) together with a PPI network analysis, in addition to novel genetic variants such as LAMA1, XYLB or NPEPL1 [60], while candidate genes such as ITGB5, RPH3A, GNAS, THY1, NEK6, JUN, GDI1, GNAI2, ERCC3 and CDC42EP4 were identified as potential biomarkers for early diagnosis in another study [61]. Gene-based multivariate tests, such as Versatile gene-based assay (VEGAS) or Multiphenotypic association analysis (MultiPhen), can also provide additional data on susceptible brain regions of AD -related functional areas [62].
1.3. Key genes as targets for AD therepies
A number of mutant variants in genes that affect the processing of APP and the formation of Aβ-peptides are considered potential targets for the development of AD therapies. One target that has been the focus of recent studies is BACE2, which plays an important role in APP-Aβ processing and cleavage. Both mutant variants at this locus and CLU expression affecting the juxta-membrane helix of APP can lead to early cleavage of this precursor protein by suppressing β-secretase activity [63]. Therapies against these targets would include prevention of CLU expression. Tauopathy associated with MAPT can be treated with drugs that stimulate microtubule stabilization by mutations at this locus or excessive phosphorylation in the brain [64]. Of note, processing and cleavage of APP into extracellular vesicles in the brain occurs with the progression of AD and contributes to both APP -dependent neurotoxicity and disease prognosis. At disease onset, vesicles are released outside brain cells along with APP and derived deleterious metabolites such as AICD and Aβ, preventing the accumulation of neurotoxic peptides in the brain [65]. This mechanism represents a potential target against AD pathogenesis, with extracellular vesicles acting protectively on brain cells.
In the sporadic form of AD, the β-secretase-derived C99 fragment is involved in the Aβ-independent aggregation of APP-CTFs in mitochondria, resulting in morphological and functional changes that can trigger pathogenic pathways in the brain. These mitochondrial abnormalities would be a target to counteract the early accumulation of APP-CTFs and slow disease progression [66]. Therapeutic treatments based on the localization of tau and Aβ proteins and APP processing must consider factors that alter their functionality, as well as changes within the cell or their expression in specific cells or body parts. TREM2 is also becoming an important target in the development of AD therapies that focus on stimulating TREM2 signaling at an early stage, before tau neuropathogenesis or deposits of β-amyloid form in the brain. However, the role of TREM2 needs to be further explored as its effect seems to change with the disease model used and the stage of tauopathy [67].
2. The Epigenome of Alzheimer's Disease as a Target for Therapeutic Treatment
Epigenetic changes can be understood as any mechanism by which the environment can alter the phenotype without altering the genotype, and they are a crucial factor that can explain the non-genetic component associated with the lack of heritability of complex traits. Epigenetic mechanisms may act as mediators of environmental factors and genetic risk components throughout an individual's life [68][69] and may play a particularly important role in the etiology of sporadic AD. Age and other factors indicative of healthy or unhealthy lifestyle habits, such as diet, smoking, and educational attainment, have been associated with the occurrence of AD [70][71][72]. Epigenetic mechanisms most commonly associated with AD include DNA methylation, post-translational modifications of histones (PTM-Hs), and gene silencing of non-coding RNAs (ncRNAs), which usually do not occur in isolation but in a complex interplay in which environmental factors may also play a role that may ultimately influence important cognitive processes [73][74]. Despite the association between many of these factors and the occurrence of epigenetic changes during the development of AD, the process that drives the interaction between each factor and leads to different rates of disease progression is not well understood. See Table 1 for recent studies of the epigenome in AD.
Table 1. Key findings from studies on the epigenetic mechanisms of Alzheimer's disease found in the NCBI PubMed database.
|
Epigenomic signal |
AD model |
Main finding |
Ref |
|
Histone acetylation |
Post-mortem human prefrontal cortices |
Tau pathology, but not amyloid-β pathology, affects histone acetylation. In neurons, altered transcription occurs due to tau-induced chromatin remodeling. Tau-related chromatin changes exhibit spatial patterns. |
[75] |
|
DNA methylation |
Post-mortem human brain tissue (AD Braak stage progression). |
Identification of genes with cell type-specific methylation signatures and documentation of age-related differential methylation dynamics in neurons (CLU, NCOR2 and SYNJ2) or glia (CXXC5, RAI1 and INPP5A). Several DNA methylation signals of neuronal (HOXA3) or glial origin (ANK1) associated with AD were validated. |
[76] |
|
DNA methylation
|
AβPPswe/PS-1 mice |
Dlg4/PSD95, a protein involved in neuronal plasticity and memory, exhibits increased expression during development regulated by histone marks that have a major impact on several processes of hippocampal plasticity and neurons. |
[77] |
|
DNA methylation
|
Postmortem human brain tissue (middle temporal gyrus) and blood samples in AD patients. |
Gene regulatory network (GRN) analysis predicts altered expression of IL6 and SIAH1 genes in brain tissues influenced by methylation and hydroxymethylation. In blood, WNT3A is the leading gene in the reference network. In both tissues, a common DMR is identified near the transcriptional starting point of the gene OXT, which encodes the neuropeptide oxytocin involved in neuromodulation of social behavior. |
[78] |
|
Histone acetylation and methylation |
Human cortical brain tissue (microglia, neuronal and oligodendrocyte) |
A subset of the variants identified by GWAS could act on super-enhancers that would affect gene expression. Knockdown of a targeted microglial enhancer carrying AD risk variants suppresses BIN1 gene expression in microglia but not in neurons or astrocytes. |
[79] |
|
DNA methylation |
DNA methylation of induced pluripotent stem cells (iPSCs) from AD patients and healthy controls. One normal cell line, one LOAD line (APOE4) and at least two EOAD cell lines (PSEN1, PSEN2) are included. |
Different DMRs of 5mC, 5hmC, and 5fC/caC are identified during differentiation of iPSCs into neurons in both normal cells and PSEN2. |
[80] |
|
DNA methylation |
Whole blood DNA from trisomy 21 (T21) patients, non-dementia patients and AD dementia patients. |
In both T21 and AD patients, at least 6 hypermethylated sites occur compared to healthy individuals. One of them is located in the ADAM10 promoter region. |
[81] |
|
DNA methylation |
Blood DNA from an international population of eleven cohorts totaling 3337 individuals. |
It has been observed that genetic factors contribute to differential DNA methylation in the hippocampus. Methylation at these sites alters the expression of genes required for hippocampal function and metabolic regulation. |
[82] |
|
mtDNA methylation
|
Postmortem PFC samples and Human cell lines HEK293T and A549. |
mtDNA methylation is negatively correlated with mitochondrial gene expression and is modulated by methyltransferase (DNMT3A), which is required for the maintenance of methylation in neurons. |
[83] |
|
DNA methylation
|
E4 and E3 mice with high fat diet (HFD) |
E4 HFD mice exhibit a unique DNA methylation profile in the hippocampus. They find that HFD-induced deficits in learning and spatial memory, but not object recognition, are more pronounced in E4 mice. |
[84] |
|
DNA methylation |
DNA methylation in the entorhinal cortex of the brain (EC) Samples from the MRC London Neurodegenerative Diseases Brain Bank. |
The ANK1 gene exhibits differential DNA methylation at AD. Abnormal DNA methylation changes in WNT5B are associated with AD neuropathology. |
[85] |
|
DNA methylation |
Peripheral blood samples from patients with MCI and normal control subjects. |
Discovery of DMRs: two within SEPT8 and TMEM232 on chromosome 5, one within SLC17A8 on chromosome 12, and another within ALOX12 on chromosome 17. Functional methylation analysis identifies four groups of genes (modules) that are significantly hypomethylated in affected individuals compared to controls: RIN3, CTSG, SPEG and UBE2L3. |
[86] |
|
Histone acetylation and methylation |
Female triple transgenic (3xTg-AD) mice |
The DNA methylation level at the promoter of the Txnip gene in the brain is significantly lower in 3xTg- AD compared to wild type. The level of DNA methylation at the CTCF region of the Txnip gene promoter is significantly lower in the cerebellum and significantly higher in the spleen of 3xTg- AD mice compared to wild-type controls. |
[87] |
|
DNA methylation |
Postmortem brains of age-matched normal controls and AD patients. |
Methylation levels in the promoter regions of the BRCA1 and AURKC genes are upregulated in AD brains. Dysfunction of BRCA1 results in impaired DNA integrity. |
[88] |
|
DNA methylation |
Four brain regions: Hippocampus, cerebellum, EC and dorsolateral PFC of donor controls and patients with late stages AD. |
Identify 858 robust DMRs in up to 772 putative genes. Identify CpGs near ANK1 and MYO1C genes with AD. The region-dependent and most significant effect is observed for a DNA methylation site near ANK1, which is more methylated in subjects with AD in the dorsolateral PFC, EC and hippocampus, but less methylated in the cerebellum. |
[89] |
|
DNA methylation |
APP/PS1 mouse |
PM20D1 is involved in lipid metabolism and is an important methylation and expression locus located within a AD -risk associated haplotype. |
[90] |
|
Histone acetylation |
THY‐Tau22 mouse |
The histone acetyltransferase (HAT)-activating molecule CBP/p300 (CSP -TTK21) is capable of acetylating nuclear chromatin in mouse brain. It shows a specific decrease in acetylation levels in the hippocampus of THY -Tau22 mice, and CSP -TTK21 significantly restores this signature by enhancing long-term spatial memory storage. |
[91] |
|
DNA methylation |
Postmortem brain tissue from patients with AD; dementia with Lewy bodies (DLB); Huntington's disease (HD); Parkinson's disease vascular dementia and non-demented control subjects. |
Significantly increased levels in AD cases compared to controls AT eight ANK1 CpG sites in the ERC and seven ANK1 CpG sites in theSTG. Changes in ANK1 DNA methylation in the cerebellum are reported for the first time. DNA hypermethylation of ANK1 in the ERC is observed only in DLB cases with coexisting AD pathology. |
[92] |
|
DNA methylation |
Postmortem hippocampal samples from AD patients and control subjects. |
DNA methylation levels correlate significantly with exposure to p-Tau deposition. Genomic loci that strongly overlap in regulatory regions are significantly hypermethylated in AD compared to healthy patients. DMGs are associated with neuronal development and neurogenesis. |
[93] |
|
Histone acetylation and methylation |
APP/PS1 mice |
Overall, no changes in histone marks over time in APP/PS1 and wild-type mice. Age-related changes in histone marks are observed in wild-type mice. |
[94] |
|
DNA methylation |
Post-mortem PFC of normal subjects and AD patients (LOAD). |
They found 504 differentially methylated positions (DMPs), of which 487 positions had increased methylation levels and 17 positions had reduced methylation levels compared to controls AD. The DMPs are mainly located in the HOXA3, GSTP1, CXXC1-3 and BIN1 genes. |
[95] |
|
DNA methylation |
Post-mortem brains of AD patients and healthy controls. |
DNA methylation (H3K9me3) is upregulated in the temporal cortex of patients with sporadic AD. |
[96] |
|
DNA methylation |
Peripheral blood monocytes from healthy controls and AD patients. Cortex samples from 4 healthy subjects and 4 AD patients. |
Hypomethylation of the TNF-α promoter region in the cerebral cortex of AD patients, while similar levels of methylation are found in control groups and in blood samples from AD patients. |
[97] |
|
DNA methylation |
Brain tissue from 147 individuals drawn from the Mount Sinai Alzheimer's and Schizophrenia Brain Bank. |
DNA hypermethylation in a 48 kb region of the HOXA gene is associated with neuropathology of AD in human cerebral cortex and cortical neuropathology of HOXA3. |
[92] |
|
DNA methylation |
DNA methylation DNA from postmortem prefrontal cortical corx tissue from patients with AD and controls. |
325 genes with differentially hydroxymethylated loci were identified. Gene enrichment analysis in ontology reveals biological processes related to the development of neuronal projections and neurogenesis. |
[98] |
|
DNA methylation |
Postmortem samples from STG patients with late onset AD and control subjects. |
A total of 17,895 CpG sites were preliminarily identified as differentially methylated between AD and control subjects, including 11,822 and 6,073 hyper- and hypomethylated CpGs, respectively. Hypermethylation was mainly detected in genes regulating cell cycle progression. |
[99] |
|
DNA methylation |
Peripheral blood (PB) samples from cognitively normal (CN), MCI, and AD patients. |
DMPs with the strongest association with MCI compared to CN are annotated with CLIP4 and those most strongly associated with AD compared to CN are annotated with FAM8A1. |
[100] |
|
DNA methylation |
LOAD patients and age- and sex-matched controls. |
Significantly higher levels of D-loop methylation are observed in heterozygous MTRR 66AG carriers compared to wild-type MTRR 66AA individuals. Stratification of the population into AD and control subgroups shows that even in the AD subgroup, carriers of the DNMT3A AA genotype have significantly higher levels of D-loop methylation than GG or GA carriers. They suggest that MTRR and DNMT3A polymorphisms affect mitochondrial DNA methylation. |
[101] |
|
Blood DNA methylation |
Blood samples from individuals before dementia diagnosis and cognitively healthy controls. |
The biggest difference in methylation is the lower methylation in diagnosed dementia compared to controls. DMRs are found together when blood samples are analysed before and after diagnosis. Genes affected by these DMRs include GULP1, SORCS3, PIEZO2, DNAH14, RIBC2, FOXG1, HOXC5, EPHA6, HOXA7, SYN3, IRX4, NOS1, LOC101929268, MARCH3, and ADAM12. |
[102] |
GWAS studies have primarily focused on specific phenotypes that include age of onset, differences in ethnicity, and psychotic traits in settings where the epigenome may influence the etiology of AD. However, effective treatment includes not only the prescription, dosage, and proper management of medications, but also the implementation of and adherence to a set of daily routines consistent with a healthy lifestyle that promotes socialization, nutrition, exercise, and mental agility [103]. Research at AD has recently aimed to highlight specific targets for disease diagnosis and treatment, including several microRNA molecules that show particular promise for the development of new therapies for neurodegenerative diseases [104][105].
2 1. Epigenomic Biomarkers
Some types of short ncRNAs, miRNAs, can interfere with the processing of Aβ and non-toxic APP via the alternative non-amyloidogenic pathway mediated by ADAM10 to form soluble APPα [106]. ADAM10 is regulated by miR-221 in AD neuroblastoma cells, and inhibition of expression of this molecule results in increased levels of the gene [107]. Another notable epigenetic marker is the mechanism of the small nuclear U1 ribonucleoprotein complex (U1-snRNP), which leads to alterations in the neuronal cell cycle via a defective RNA splicing process and ultimately affects the metabolic and biochemical processes responsible for neuroinflammation, cell decay and death [108][109]. Putative epigenomic biomarkers such as miR-129, which is thought to be ubiquitously upregulated in AD brains, may furthermore be useful for drug treatment against target genes [110].
2.2. Aβ Immunotherapy
Treatments based on Aβ immunotherapy appear to be effective in clearing amyloid plaques in the human brain, but not in slowing disease progression or reversing cognitive impairment. The use of immune checkpoint blockers (ICBs) is gaining interest as a more effective epigenetic target against the neuroinflammatory processes that characterize AD amyloid pathology [108]. Some ICBs based on antibodies against the protein ligand 1 complex of programmed cell death (PD-L1) are being investigated as therapies for cancer. In AD animal models, the use of drugs targeting the PD-1/PD-L1 complex can trigger an immune response that prevents the accumulation of neurotoxic substances associated with APP [111]. Inhibition of this complex promotes ligand degradation in antigen-presenting cells by increasing immune tolerance and preventing T-cell degradation, which helps to reduce neuroinflammation and recover from impaired cognitive functions [112][113]. However, the results of various experiments with BCIs are conflicting regarding their therapeutic ability to treat AD [114][115], and further research in this area is needed.
2.3. HDAC Inhibitors
Targeted therapy with HDAC inhibitors (HDACis) aims to reduce the cognitive deficits associated with AD or other neuropathologies [116][117]. The US Food and Drug Administration (FDA) and the Chinese agency of the same name (CFDA) have approved some HDACi drugs such as vorinostat (SAHA), panobinostat (LBH589), belinostat (PXD101), romidepsin (FK-228), and chidamide (HBI-8000), and although most of them were primarily developed to treat hematologic malignancies, some are also being investigated for the treatment of CNS disorders [118]. Lacosamide [119], tubastatin A [120], quisinostat [121], trichostatin A [122], and M344 [123] are the other HDACi that have recently been reported as prominent targets for AD. Valproic acid [124], 4-phenylbutyrate [125], MPT0G211 [126] and nicotinamide [127] also showed similar therapeutic effects in AD animal models. By combining the crucial structural features of the antioxidant ebselen and HDACi pharmacophores (vorinostat, tubastatin A, panobinostat and quisinostat), a class of novel synthetic hybrid compounds was created for AD therapy, and the compound identified as 7f was a potent HDACi [128]. The efficacy of the compounds CM-414 and CM-695 as a novel multitarget therapy focusing on inhibition of HDACs and phosphodiesterase 5 (PDE5) was demonstrated in Tg2576 mice, showing inhibition of intermediate class I HDACs and greater inhibition of HDAC6 and PDE5 [129][130]. Finally, Lim et al. pioneered the development of novel aspirin-inspired acetyl donor HDACi [118].
References
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867.
- Panegyres, P.K.; Chen, H.-Y. Differences between Early and Late Onset Alzheimer’s Disease. Am. J. Neurodegener. Dis. 2013, 2, 300–306.
- Mendez, M.F. Early-Onset Alzheimer Disease. Neurol. Clin. 2017, 35, 263–281.
- Braak, H.; Del Tredici, K.; Braak, H.; Del Tredici, K. The Pathological Process Underlying Alzheimer’s Disease in Individuals under Thirty. Acta Neuropathol 121: 171-181. Acta Neuropathol. 2011, 121, 171–181.
- Gatz, M.; Pedersen, N.L. Study of Dementia in Swedish Twins. Twin Res. Hum. Genet. 2013, 16, 313–316.
- Yokoyama, A.; Rutledge, J.; Medici, V. DNA Methylation Alterations in Alzheimer’s Disease. Environ. Epigenetics 2017, 3, dvx008.
- D’addario, C.; Bastías-Candia, S.; Arosio, B.; Di Bartolomeo, M.; Abbate, C.; Casè, A.; Candeletti, S.; Romualdi, P.; Damanti, S.; Maccarrone, M.; et al. Transcriptional and Epigenetic Phenomena in Peripheral Blood Cells of Monozygotic Twins Discordant for Alzheimer’s Disease, a Case Report. J. Neurol. Sci. 2016, 372, 211–216.
- Levy, E.; Carman, M.; Fernandez-Madrid, I.J.; Power, M.; Lieberburg, I.; Duinen, S.G.; Bots, G.; Luyendijk, W.; Frangione, B. Mutation of the Alzheimer’s Disease Amyloid Gene in Hereditary Cerebral Hemorrhage, Dutch Type. Science 1990, 248, 1124–1126.
- Rogaev, E.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s Disease in Kindreds with Missense Mutations in a Gene on Chromosome 1 Related to the Alzheimer’s Disease Type 3 Gene. Nature 1995, 376, 775–778.
- Genin, E.; Hannequin, D.; Wallon, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Bullido, M.; Engelborghs, S.; Paul, D.; Berr, C.; et al. APOE and Alzheimer Disease: A Major Gene with Semi-Dominant Inheritance. Mol. Psychiatry 2011, 16, 903–907.
- McKay, G.J.; Silvestri, G.; Chakravarthy, U.; Dasari, S.; Fritsche, L.G.; Weber, B.H.; Keilhauer, C.N.; Klein, M.L.; Francis, P.J.; Klaver, C.C.; et al. Variations in Apolipoprotein E Frequency with Age in a Pooled Analysis of a Large Group of Older People. Am. J. Epidemiol. 2011, 173, 1357–1364.
- Martínez-Magaña, J.; Genis-Mendoza, A.; Tovilla-Zarate, C.A.; González-Castro, T.; Juárez-Rojop, I.; Hernández-Díaz, Y.; Martínez-H, A.; García-Ortiz, H.; Orozco, L.; Narváez, L.; et al. Association between APOE Polymorphisms and Lipid Profile in Mexican Amerindian Population. Mol. Genet. Genom. Med. 2019, 7, e958.
- Keeney, J.; Ibrahimi, S.; Zhao, L. Human ApoE Isoforms Differentially Modulate Glucose and Amyloid Metabolic Pathways in Female Brain: Evidence of the Mechanism of Neuroprotection by ApoE2 and Implications for Alzheimer’s Disease Prevention and Early Intervention. J. Alzheimers. Dis. 2015, 48, 411–424.
- Neu, S.C.; Pa, J.; Kukull, W.; Beekly, D.; Kuzma, A.; Gangadharan, P.; Wang, L.-S.; Romero, K.; Arneric, S.P.; Redolfi, A.; et al. Apolipoprotein E Genotype and Sex Risk Factors for Alzheimer Disease: A Meta-Analysis. JAMA Neurol. 2017, 74, 1178–1189.
- Escott-Price, V.; Shoai, M.; Pither, R.; Williams, J.; Hardy, J. Polygenic Score Prediction Captures Nearly All Common Genetic Risk for Alzheimer’s Disease. Neurobiol. Aging 2016, 49, 214.e7–214.e11.
- Gatz, M.; Reynolds, C.; Fratiglioni, L.; Johansson, B.; Mortimer, J.; Berg, S.; Fiske, A.; Pedersen, N. Role of Genes and Environments for Explaining Alzheimer Disease. Arch. Gen. Psychiatry 2006, 63, 168–174.
- Zhang, S.; Cai, F.; Wu, Y.; Bozorgmehr, T.; Wang, Z.; Zhang, S.; Huang, D. A Presenilin-1 Mutation Causes Alzheimer Disease without Affecting Notch Signaling. Mol. Psychiatry 2020, 25, 603–613.
- Barber, I.; Braae, A.; Clement, N.; Patel, T.; Guetta-Baranes, T.; Brookes, K.-J.; Medway, C.; Chappell, S.; Guerreiro, R.; Bras, J.; et al. Mutation Analysis of Sporadic Early-Onset Alzheimer’s Disease Using the NeuroX Array. Neurobiol. Aging 2016, 49, 215.e1–215.e8.
- Lanoiselee, H.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 Mutations in Early-Onset Alzheimer Disease: A Genetic Screening Study of Familial and Sporadic Cases. PLoS Med. 2017, 14, e1002270.
- Giaccone, G.; Morbin, M.; Moda, F.; Botta, M.; Mazzoleni, G.; Uggetti, A.; Catania, M.; Redaelli, V.; Spagnoli, A.; Rossi, R.; et al. Neuropathology of the Recessive A673V APP Mutation: Alzheimer Disease with Distinctive Features. Acta Neuropathol. 2010, 120, 803–812.
- Zhang, S.; Wang, Z.; Cai, F.; Zhang, M.; Wu, Y.; Zhang, J.; Song, W. BACE1 Cleavage Site Selection Critical for Amyloidogenesis and Alzheimer’s Pathogenesis. J. Neurosci. 2017, 37, 317–340.
- Holler, C.J.; Davis, P.R.; Beckett, T.L.; Platt, T.L.; Webb, R.L.; Head, E.; Murphy, M.P. Bridging Integrator 1 (BIN1) Protein Expression Increases in the Alzheimer’s Disease Brain and Correlates with Neurofibrillary Tangle Pathology. J. Alzheimers. Dis. 2014, 42, 1221–1227.
- Franzmeier, N.; Rubinski, A.; Neitzel, J.; Ewers, M.; Weiner, M.W.; Aisen, P.; Petersen, R.; Jack, C.R.; Jagust, W.; Trojanowski, J.Q.; et al. The BIN1 Rs744373 SNP Is Associated with Increased Tau-PET Levels and Impaired Memory. Nat. Commun. 2019, 10, 1766.
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P.; et al. Increased Expression of BIN1 Mediates Alzheimer Genetic Risk by Modulating Tau Pathology. Mol. Psychiatry 2013, 18, 1225–1234.
- Lacour, A.; Espinosa, A.; Louwersheimer, E.; Heilmann, S.; Hernández, I.; Wolfsgruber, S.; Fernández, V.; Wagner, H.; Rosende-Roca, M.; Mauleón, A.; et al. Genome-Wide Significant Risk Factors for Alzheimer’s Disease: Role in Progression to Dementia Due to Alzheimer’s Disease among Subjects with Mild Cognitive Impairment. Mol. Psychiatry 2017, 22, 153–160.
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.-S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common Variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 Are Associated with Late-Onset Alzheimer’s Disease. Nat. Genet. 2011, 43, 436–441.
- Kang, S.; Kurti, A.; Wojtas, A.; Baker, K.; Liu, C.-C.; Kanekiyo, T.; Deming, Y.; Cruchaga, C.; Estus, S.; Bu, G.; et al. Identification of Plexin A4 as a Novel Clusterin Receptor Links Two Alzheimer’s Disease Risk Genes. Hum. Mol. Genet. 2016, 25, ddw188.
- Tan, L.; Wang, H.-F.; Tan, M.-S.; Tan, C.-C.; Zhu, X.-C.; Miao, D.; Yu, W.-J.; Jiang, T.; Tan, L.; Yu, J.-T.; et al. Effect of CLU Genetic Variants on Cerebrospinal Fluid and Neuroimaging Markers in Healthy, Mild Cognitive Impairment and Alzheimer’s Disease Cohorts. Sci. Rep. 2016, 6, 26027.
- Bailey, C.; DeVaux, L.; Farzan, M. The Triggering Receptor Expressed on Myeloid Cells 2 Binds Apolipoprotein E. J. Biol. Chem. 2015, 290, 26033–26042.
- Hoeijmakers, L.; Ruigrok, S.R.; Amelianchik, A.; Ivan, D.; van Dam, A.-M.; Lucassen, P.J.; Korosi, A. Early-Life Stress Lastingly Alters the Neuroinflammatory Response to Amyloid Pathology in an Alzheimer’s Disease Mouse Model. Brain. Behav. Immun. 2017, 63, 160–175.
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons From Other Pathologies. Front. Neurosci. 2019, 13, 164.
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-Transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Aβ and Synaptic Dysfunction. Neuron 2003, 39, 409–421.
- Kumita, J.; Poon, S.; Caddy, G.; Hagan, C.; Dumoulin, M.; Yerbury, J.; Stewart, E.; Robinson, C.; Wilson, M.; Dobson, C. The Extracellular Chaperone Clusterin Potently Inhibits Human Lysozyme Amyloid Formation by Interacting with Prefibrillar Species. J. Mol. Biol. 2007, 369, 157–167.
- Wojtas, A.M.; Kang, S.S.; Olley, B.M.; Gatherer, M.; Shinohara, M.; Lozano, P.A.; Liu, C.-C.; Kurti, A.; Baker, K.E.; Dickson, D.W.; et al. Loss of Clusterin Shifts Amyloid Deposition to the Cerebrovasculature via Disruption of Perivascular Drainage Pathways. Proc. Natl. Acad. Sci. USA 2017, 114, E6962–E6971.
- Xiao, Q.; Gil, S.-C.; Yan, P.; Wang, Y.; Han, S.; Gonzales, E.; Perez, R.; Cirrito, J.; Lee, J.-M. Role of Phosphatidylinositol Clathrin Assembly Lymphoid-Myeloid Leukemia (PICALM) in Intracellular Amyloid Precursor Protein (APP) Processing and Amyloid Plaque Pathogenesis. J. Biol. Chem. 2012, 287, 21279–21289.
- Zhou, Q.; Peng, D.; Yuan, X.; Lv, Z.; Pang, S.; Jiang, W.; Yang, C.; Shi, X.; Pang, G.; Yang, Y.; et al. APOE and APOC1 Gene Polymorphisms Are Associated with Cognitive Impairment Progression in Chinese Patients with Late-Onset Alzheimer’s Disease. Neural Regen. Res. 2014, 9, 653–660.
- Vardarajan, B.; Zhang, Y.; Lee, J.; Cheng, R.; Bohm, C.; Ghani, M.; Reitz, C.; Reyes-Dumeyer, D.; Shen, Y.; Rogaeva, E.; et al. Coding Mutations in SORL1 and Alzheimer Disease. Ann. Neurol. 2014, 77, 215–227.
- Li, X.; Zhu, X.; Zhang, W.; Yang, F.; Hui, J.; Tan, J.; Xie, H.; Peng, D.; Ma, L.; Cui, L.; et al. The Etiological Effect of a New Low-Frequency ESR1 Variant on Mild Cognitive Impairment and Alzheimer’s Disease: A Population-Based Study. Aging 2018, 10, 2316–2337.
- Cuccaro, M.; Carney, R.; Zhang, Y.; Bohm, C.; Kunkle, B.; Vardarajan, B.; Whitehead, P.; Cukier, H.; Mayeux, R.; George-Hyslop, P.; et al. SORL1 Mutations in Early- and Late-Onset Alzheimer Disease. Neurol. Genet. 2016, 2, e116.
- Miller, J.; Woltjer, R.; Goodenbour, J.; Horvath, S.; Geschwind, D. Genes and Pathways Underlying Regional and Cell Type Changes in Alzheimer’s Disease. Genome Med. 2013, 5, 48.
- Blalock, E.; Buechel, H.; Popovic, J.; Geddes, J.; Landfield, P. Microarray Analyses of Laser-Captured Hippocampus Reveal Distinct Gray and White Matter Signatures Associated with Incipient Alzheimer’s Disease. J. Chem. Neuroanat. 2011, 42, 118–126.
- Engelhart, M.; Geerlings, M.; Meijer, J.; Kiliaan, A.; Ruitenberg, A.; Swieten, J.; Stijnen, T.; Hofman, A.; Witteman, J.; Breteler, M. Inflammatory Proteins in Plasma and the Risk of Dementia: The Rotterdam Study. Arch. Neurol. 2004, 61, 668–672.
- Padmanabhan, J.; Judge, M.; Dickson, D.; Potter, H. α 1-Antichymotrypsin, an Inflammatory Protein Overexpressed in Alzheimer’s Disease Brain, Induces Tau Phosphorylation in Neurons. Brain 2006, 129, 3020–3034.
- Kamboh, M.I.; Minster, R.L.; Kenney, M.; Ozturk, A.; Desai, P.P.; Kammerer, C.M.; DeKosky, S.T. α-1-Antichymotrypsin (ACT or SERPINA3) Polymorphism May Affect Age-at-Onset and Disease Duration of Alzheimer’s Disease. Neurobiol. Aging 2006, 27, 1435–1439.
- Nilsson, L.N.; Bales, K.R.; DiCarlo, G.; Gordon, M.N.; Morgan, D.; Paul, S.M.; Potter, H. α-1-Antichymotrypsin Promotes β-Sheet Amyloid Plaque Deposition in a Transgenic Mouse Model of Alzheimer’s Disease. J. Neurosci. 2001, 21, 1444–1451.
- Jonsson, T.; Stefansson, H.; Steinberg, S.; Jonsdottir, I.; Jonsson, P.; Snaedal, J.; Bjornsson, S.; Huttenlocher, J.; Levey, A.; Lah, J.; et al. Variant of TREM2 Associated with the Risk of Alzheimer’s Disease. N. Engl. J. Med. 2013, 368, 107–116.
- Slattery, C.F.; Beck, J.A.; Harper, L.; Adamson, G.; Abdi, Z.; Uphill, J.; Campbell, T.; Druyeh, R.; Mahoney, C.J.; Rohrer, J.D.; et al. R47H TREM2 Variant Increases Risk of Typical Early-Onset Alzheimer’s Disease but Not of Prion or Frontotemporal Dementia. Alzheimer’s Dement. 2014, 10, 602–608.e4.
- Lill, C.; Rengmark, A.; Pihlstrøm, L.; Fogh, I.; Shatunov, A.; Sleiman, P.; Wang, L.-S.; Liu, T.; Funch Lassen, C.; Meissner, E.; et al. The Role of TREM2 R47H as a Risk Factor for Alzheimer’s Disease, Frontotemporal Lobar Degeneration, Amyotrophic Lateral Sclerosis, and Parkinson’s Disease. Alzheimers. Dement. 2015, 11, 1407–1416.
- Vo, G.; Bagyinszky, E.; Yang, Y.; Youn, Y.C.; An, S.; Kim, S. Genetic Analyses of Early-Onset Alzheimer’s Disease Using next-Generation Sequencing. Sci. Rep. 2019, 9, 8368.
- Cochran, J.; Rush, T.; Buckingham, S.; Roberson, E. The Alzheimer’s Disease Risk Factor CD2AP Maintains Blood-Brain Barrier Integrity. Hum. Mol. Genet. 2015, 24, 6667–6674.
- Tao, Q.-Q.; Zhijun, L.; Sun, Y.-M.; Li, H.-L.; Yang, P.; Jiang, B.; Li, X.-Y.; Xu, J.-F.; Wu, Z.-Y. Decreased Gene Expression of CD2AP in Chinese Patients with Sporadic Alzheimer’s Disease. Neurobiol. Aging 2017, 56, 212.e5–212.e10.
- Allen, M.; Kachadoorian, M.; Quicksall, Z.; Zou, F.; Chai, H.; Younkin, C.; Crook, J.; Pankratz, V.; Carrasquillo, M.; Krishnan, S.; et al. Association of MAPT Haplotypes with Alzheimer’s Disease Risk and MAPT Brain Gene Expression Levels. Alzheimers. Res. Ther. 2014, 6, 39.
- Soyal, S.M.; Kwik, M.; Kalev, O.; Lenz, S.; Zara, G.; Strasser, P.; Patsch, W.; Weis, S. A TOMM40/APOE Allele Encoding APOE-E3 Predicts High Likelihood of Late-Onset Alzheimer’s Disease in Autopsy Cases. Mol. Genet. Genom. Med. 2020, 8, e1317.
- Nelson, P.; Alafuzoff, I.; Bigio, E.; Bouras, C.; Braak, H.; Cairns, N.; Castellani, R.; Crain, B.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381.
- Vassar, R. ADAM10 Prodomain Mutations Cause Late-Onset Alzheimer’s Disease: Not Just the Latest FAD. Neuron 2013, 80, 250–253.
- Atagi, Y.; Liu, C.-C.; Painter, M.M.; Chen, X.-F.; Verbeeck, C.; Zheng, H.; Li, X.; Rademakers, R.; Kang, S.S.; Xu, H.; et al. Apolipoprotein E Is a Ligand for Triggering Receptor Expressed on Myeloid Cells 2 (TREM2). J. Biol. Chem. 2015, 290, 26043–26050.
- Ebbert, M.T.W.; Boehme, K.L.; Wadsworth, M.E.; Staley, L.A.; Initiative, A.D.N.; Consortium, A.D.G.; Mukherjee, S.; Crane, P.K.; Ridge, P.G.; Kauwe, J.S.K. Interaction between Variants in CLU and MS4A4E Modulates Alzheimer’s Disease Risk. Alzheimers. Dement. 2016, 12, 121–129.
- Goñi, J.; Esteban, F.; Velez de Mendizabal, N.; Sepulcre, J.; Ardanza-Trevijano, S.; Agirrezabal, I.; Villoslada, P. A Computational Analysis of Protein-Protein Interaction Networks in Neurodegenerative Diseases. BMC Syst. Biol. 2008, 2, 52.
- Marín, M.; Esteban, F.; Ramirez, H.; Ros, E.; Saez-Lara, M. An Integrative Methodology Based on Protein-Protein Interaction Networks for Identification and Functional Annotation of Disease-Relevant Genes Applied to Channelopathies. BMC Bioinform. 2019, 20, 565.
- Meng, X.; Li, J.; Zhang, Q.; Chen, F.; Bian, C.; Yao, X.; Yan, J.; Xu, Z.; Risacher, S.; Saykin, A.; et al. Multivariate Genome Wide Association and Network Analysis of Subcortical Imaging Phenotypes in Alzheimer’s Disease. BMC Genom. 2020, 21, 896.
- Wu, M.; Fang, K.; Wang, W.; Lin, W.; Guo, L.; Wang, J. Identification of Key Genes and Pathways for Alzheimer’s Disease via Combined Analysis of Genome-Wide Expression Profiling in the Hippocampus. Biophys. Rep. 2019, 5, 98–109.
- Chung, J.; Jun, G.; Dupuis, J.; Farrer, L. Comparison of Methods for Multivariate Gene-Based Association Tests for Complex Diseases Using Common Variants. Eur. J. Hum. Genet. 2019, 27, 811–823.
- Wang, Z.; Xu, Q.; Cai, F.; Liu, X.; Wu, Y.; Song, W. BACE2, a Conditional β-Secretase, Contributes to Alzheimer’s Disease Pathogenesis. JCI Insight 2019, 4, e123431.
- Varidaki, A.; Hong, Y.; Coffey, E.T. Repositioning Microtubule Stabilizing Drugs for Brain Disorders. Front. Cell. Neurosci. 2018, 12, 226.
- Perez-Gonzalez, R.; Kim, Y.; Miller, C.; Pacheco-Quinto, J.; Eckman, E.; Levy, E. Extracellular Vesicles: Where the Amyloid Precursor Protein Carboxyl-Terminal Fragments Accumulate and Amyloid-β Oligomerizes. FASEB J. 2020, 34, 12922–12931.
- Vaillant-Beuchot, L.; Mary, A.; Pardossi-Piquard, R.; Bourgeois, A.; Lauritzen, I.; Eysert, F.; Kinoshita, P.F.; Cazareth, J.; Badot, C.; Fragaki, K.; et al. Accumulation of Amyloid Precursor Protein C-terminal Fragments Triggers Mitochondrial Structure, Function, and Mitophagy Defects in Alzheimer’s Disease Models and Human Brains. Acta Neuropathol. 2021, 141, 39–65.
- Gratuze, M.; Leyns, C.; Holtzman, D. New Insights into the Role of TREM2 in Alzheimer’s Disease. Mol. Neurodegener. 2018, 13, 1–6.
- Aristizabal, M.; Anreiter, I.; Halldorsdottir, T.; Odgers, C.; Mcdade, T.; Goldenberg, A.; Mostafavi, S.; Kobor, M.; Binder, E.;Sokolowski, M.; et al. Biological Embedding of Experience: A Primer on Epigenetics. Proc. Natl. Acad. Sci. USA2019, 117, 23261–23269. [CrossRef]
- Li, S.; Nguyen, T.; Wong, E.; Dugué, P.-A.; Dite, G.; Armstrong, N.; Craig, J.; Mather, K.; Sachdev, P.; Saffery, R.; et al. Genetic and Environmental Causes of Variation in Epigenetic Aging across the Lifespan. Clin. Epigenetics 2020, 12, 158.
- Tsang, V.; Fry, R.; Niculescu, M.; Rager, J.; Saunders, J.; Paul, D.; Zeisel, S.; Waalkes, M.; Stýblo, M.; Drobná, Z. The Epigenetic Effects of a High Prenatal Folate Intake in Male Mouse Fetuses Exposed In Utero to Arsenic. Toxicol. Appl. Pharmacol.2012, 264, 439–450.
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.;Peters, A.; et al. Tobacco Smoking Leads to Extensive Genome-Wide Changes in DNA Methylation. PLoS ONE 2013, 8, e63812.
- Ambatipudi, S.; Cuenin, C.; Hernández-Vargas, H.; Ghantous, A.; Calvez-Kelm, F.; Kaaks, R.; Barrdahl, M.; Boeing, H.; Aleksandrova, K.; Trichopoulou, A.; et al. Tobacco Smoking-Associated Genome-Wide DNA Methylation Changes in the EPIC Study. Epigenomics 2016, 8, 599–618.
- Yoon, H.-G.; Chan, D.; Reynolds, A.; Qin, J.; Wong, J. N-CoR Mediates DNA Methylation-Dependent Repression through a Methyl CpG Binding Protein Kaiso. Mol. Cell 2003, 12, 723–734.
- Rottach, A.; Frauer, C.; Pichler, G.; Bonapace, I.; Spada, F.; Leonhardt, H. The Multi-Domain Protein Np95 Connects DNA Methylation and Histone Modification. Nucleic Acids Res. 2009, 38, 1796–1804.
- Hans-Ulrich Klein; Cristin McCabe; Elizabeta Gjoneska; Sarah E. Sullivan; Belinda J. Kaskow; Anna Tang; Robert V. Smith; Jishu Xu; Andreas R. Pfenning; Bradley E. Bernstein; et al.Alexander MeissnerJulie A. SchneiderSara MostafaviLi-Huei TsaiTracy L. Young-PearseDavid A. BennettPhilip L. De Jager Epigenome-wide study uncovers large-scale changes in histone acetylation driven by tau pathology in aging and Alzheimer’s human brains. Nature Neuroscience 2018, 22, 37-46, 10.1038/s41593-018-0291-1.
- Gilles Gasparoni; Sebastian Bultmann; Pavlo Lutsik; Theo F. J. Kraus; Sabrina Sordon; Julia Vlcek; Vanessa Dietinger; Martina Steinmaurer; Melanie Haider; Christopher B. Mulholland; et al.Thomas ArzbergerSigrun RoeberMatthias RiemenschneiderHans A. KretzschmarArmin GieseHeinrich LeonhardtJörn Walter DNA methylation analysis on purified neurons and glia dissects age and Alzheimer’s disease-specific changes in the human cortex. Epigenetics & Chromatin 2018, 11, 1-19, 10.1186/s13072-018-0211-3.
- Fernando Bustos; Estibaliz Ampuero; Nur Jury; Rodrigo Aguilar; Fahimeh Falahi; Jorge Toledo; Juan Ahumada; Jaclyn Lata; Paula Cubillos; Berta Henríquez; et al.Miguel V GuerraJimmy StehbergRachael L NeveNibaldo C InestrosaUrsula WynekenMarco FuenzalidaSteffen HärtelMiguel Sena-EstevesLorena Varela-NallarMarianne G RotsMartin MontecinoBrigitte Van Zundert Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain 2017, 140, 3252-3268, 10.1093/brain/awx272.
- Roy Lardenoije; Janou A. Y. Roubroeks; Ehsan Pishva; Markus Leber; Holger Wagner; Artemis Iatrou; Adam Smith; Rebecca G. Smith; Lars M. T. Eijssen; Luca Kleineidam; et al.Amit KawaliaPer HoffmannTobias LuckSteffi Riedel-HellerFrank JessenWolfgang MaierMichael WagnerRene HurlemannGunter KenisMuhammad AliAntonio Del SolDiego MastroeniElaine DelvauxPaul D. ColemanJonathan MillBart P. F. RuttenKatie LunnonAlfredo RamirezDaniël L. A. Van Den Hove Alzheimer’s disease-associated (hydroxy)methylomic changes in the brain and blood. Clinical Epigenetics 2019, 11, 164, 10.1186/s13148-019-0755-5.
- Alexi Nott; Inge R. Holtman; Nicole G. Coufal; Johannes C. M. Schlachetzki; Miao Yu; Rong Hu; Claudia Z. Han; Monique Pena; Jiayang Xiao; Yin Wu; et al.Zahara KeulenMartina P. PasillasCarolyn O’ConnorChristian K. NicklSimon T. SchaferZeyang ShenRobert A. RissmanJames B. BrewerDavid GosselinDavid D. GondaMichael L. LevyMichael G. RosenfeldGraham McVickerFred H. GageBing RenChristopher K. Glass Brain cell type–specific enhancer–promoter interactome maps and disease - risk association. Science 2019, 366, 1134-1139, 10.1126/science.aay0793.
- Irfete S. Fetahu; Dingailu Ma; Kimberlie Rabidou; Christian Argueta; Michael Smith; Hang Liu; Feizhen Wu; Yujiang G. Shi; Epigenetic signatures of methylated DNA cytosine in Alzheimer’s disease. Science Advances 2019, 5, eaaw2880, 10.1126/sciadv.aaw2880.
- Larissa Haertle; Tobias Müller; Roy Lardenoije; Anna Maierhofer; Marcus Dittrich; Renzo J. M. Riemens; Samantha Stora; Mathilde Roche; Markus Leber; Steffi Riedel-Heller; et al.Michael WagnerMartin SchererAimé RavelClotilde MircherCecile Cieuta-WaltiSophie DurandDaniel L. A. Van De HovePer HoffmannAlfredo RamirezThomas HaafNady El HajjAndré Mégarbané Methylomic profiling in trisomy 21 identifies cognition- and Alzheimer’s disease-related dysregulation. Clinical Epigenetics 2019, 11, 1-11, 10.1186/s13148-019-0787-x.
- Tianye Jia; Congying Chu; Yun Liu; Jenny van Dongen; Evangelos Papastergios; Nicola J. Armstrong; Mark E. Bastin; Tania Carrillo-Roa; Anouk Den Braber; Mathew Harris; et al.Rick JansenJingyu LiuMichelle LucianoAnil P. S. OriRoberto Roiz SantiañezBarbara RuggeriDaniil SarkisyanJean ShinKim SungeunDiana Tordesillas GutiérrezDennis Van’T EntDavid AmesEric ArtigesGeorgy BakalkinTobias BanaschewskiArun L. W. BokdeHenry BrodatyUli BrombergRachel BrouwerChristian BüchelErin Burke QuinlanWiepke CahnGreig I. de ZubicarayStefan EhrlichTomas J. EkströmHerta FlorJuliane H. FröhnerVincent FrouinHugh GaravanPenny GowlandAndreas HeinzJacqueline HoareBernd IttermannNeda JahanshadJiyang JiangJohn B. KwokNicholas G. MartinJean-Luc MartinotKaren A. MatherKatie L. McMahonAllan F. McRaeFrauke NeesDimitri Papadopoulos OrfanosTomáš PausLuise PoustkaPhilipp G. SämannPeter R. SchofieldMichael N. SmolkaDan J. SteinLachlan T. StrikeJalmar TeeuwAnbupalam ThalamuthuJulian TrollorHenrik WalterJoanna M. WardlawWei WenRobert WhelanLiana G. ApostolovaElisabeth B. BinderDorret I. BoomsmaVince CalhounBenedicto Crespo-FacorroIan J. DearyHilleke Hulshoff PolRoel A. OphoffZdenka PausovaPerminder S. SachdevAndrew SaykinMargaret J. WrightPaul M. ThompsonGunter SchumannSylvane Desrivières Epigenome-wide meta-analysis of blood DNA methylation and its association with subcortical volumes: findings from the ENIGMA Epigenetics Working Group. Molecular Psychiatry 2019, 26, 3884-3895, 10.1038/s41380-019-0605-z.
- Xiaoyang Dou; Jerome D. Boyd-Kirkup; Joseph McDermott; Xiaoli Zhang; Fang Li; Bowen Rong; Rui Zhang; Bisi Miao; Peilin Chen; Hao Cheng; et al.Jianhuang XueDavid BennettJiemin WongFei LanJing-Dong J. Han The strand-biased mitochondrial DNA methylome and its regulation by DNMT3A. Genome Research 2019, 29, 1622-1634, 10.1101/gr.234021.117.
- Lance A. Johnson; Eileen Ruth S. Torres; Soren Impey; Jan Frederik Stevens; Jacob Raber; Apolipoprotein E4 and Insulin Resistance Interact to Impair Cognition and Alter the Epigenome and Metabolome. Scientific Reports 2017, 7, srep43701, 10.1038/srep43701.
- Adam R. Smith; Rebecca G. Smith; Joe Burrage; Claire Troakes; Safa Al-Sarraj; Rajesh N. Kalaria; Carolyn Sloan; Andrew Robinson; Jonathan Mill; Katie Lunnon; et al. A cross-brain regions study of ANK1 DNA methylation in different neurodegenerative diseases. Neurobiology of Aging 2018, 74, 70-76, 10.1016/j.neurobiolaging.2018.09.024.
- Gita A. Pathak; Talisa K. Silzer; Jie Sun; Zhengyang Zhou; Ann A. Daniel; Leigh Johnson; Sid O’Bryant; Nicole R. Phillips; Robert C. Barber; Genome-Wide Methylation of Mild Cognitive Impairment in Mexican Americans Highlights Genes Involved in Synaptic Transport, Alzheimer’s Disease-Precursor Phenotypes, and Metabolic Morbidities. Journal of Alzheimer's Disease 2019, 72, 733-749, 10.3233/JAD-190634.
- Emre Fertan; Gloria J. Rodrigues; Ryan V. Wheeler; Donna Goguen; Aimee A. Wong; Hana James; Andrew Stadnyk; Richard E. Brown; Ian C.G. Weaver; Cognitive Decline, Cerebral-Spleen Tryptophan Metabolism, Oxidative Stress, Cytokine Production, and Regulation of the Txnip Gene in a Triple Transgenic Mouse Model of Alzheimer Disease. The American Journal of Pathology 2019, 189, 1435-1450, 10.1016/j.ajpath.2019.03.006.
- Tatsuo Mano; Kenichi Nagata; Takashi Nonaka; Airi Tarutani; Tomohiro Imamura; Tadafumi Hashimoto; Taro Bannai; Kagari Koshi-Mano; Takeyuki Tsuchida; Ryo Ohtomo; et al.Junko Takahashi-FujigasakiSatoshi YamashitaYasumasa OhyagiRyo YamasakiShoji TsujiAkira TamaokaTakeshi IkeuchiTakaomi C. SaidoTakeshi IwatsuboToshikazu UshijimaShigeo MurayamaMasato HasegawaAtsushi Iwata Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proceedings of the National Academy of Sciences 2017, 114, E9645-E9654, 10.1073/pnas.1707151114.
- Stephen A. Semick; Rahul A. Bharadwaj; Leonardo Collado-Torres; Ran Tao; Joo Heon Shin; Amy Deep-Soboslay; James Weiss; Daniel R. Weinberger; Thomas M. Hyde; Joel E. Kleinman; et al.Andrew E. JaffeVenkata S. Mattay Integrated DNA methylation and gene expression profiling across multiple brain regions implicate novel genes in Alzheimer’s disease. Acta neuropathologica 2018, 137, 557–569, 10.1101/430603.
- Jose V. Sanchez-Mut; Holger Heyn; Bianca A. Silva; Lucie Dixsaut; Paula Garcia Esparcia; Enrique Vidal; Sergi Sayols; Liliane Glauser; Ana Monteagudo-Sánchez; Jordi Perez-Tur; et al.Isidre FerrerDavid MonkBernard SchneiderManel EstellerJohannes Gräff PM20D1 is a quantitative trait locus associated with Alzheimer’s disease. Nature Medicine 2018, 24, 598-603, 10.1038/s41591-018-0013-y.
- Snehajyoti Chatterjee; Raphaelle Cassel; Anne Schneider‐Anthony; Karine Merienne; Brigitte Cosquer; Laura Tzeplaeff; Sarmistha Halder Sinha; Manoj Kumar; Piyush Chaturbedy; Muthusamy Eswaramoorthy; et al.Stéphanie Le GrasCéline KeimeOlivier BousigesPatrick DutarPetnoi PetsophonsakulClaire RamponJean‐Christophe CasselLuc BueeDavid BlumTapas K KunduAnne‐Laurence Boutillier Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Molecular Medicine 2018, 10, e8587, 10.15252/emmm.201708587.
- Adam Smith; Rebecca G. Smith; Ehsan Pishva; Eilis Hannon; Janou A. Y. Roubroeks; Joe Burrage; Claire Troakes; Safa Al-Sarraj; Carolyn Sloan; Jonathan Mill; et al.Daniel Van Den HoveKatie Lunnon Parallel profiling of DNA methylation and hydroxymethylation highlights neuropathology-associated epigenetic variation in Alzheimer’s disease. Clinical Epigenetics 2019, 11, 1-13, 10.1186/s13148-019-0636-y.
- Miren Altuna; Amaya Urdánoz Casado; Javier Sánchez-Ruiz de Gordoa; María V. Zelaya; Alberto Labarga; Julie M. J. Lepesant; Miren Roldán; Idoia Blanco-Luquin; Álvaro Perdones; Rosa Larumbe; et al.Ivonne JericóCarmen EchavarriIván Méndez-LópezLuisa Di StefanoMaite Mendioroz DNA methylation signature of human hippocampus in Alzheimer’s disease is linked to neurogenesis. Clinical Epigenetics 2019, 11, 1-16, 10.1186/s13148-019-0672-7.
- Marcus Dyer; Andrew Phipps; Stanislaw Mitew; Phillippa C. Taberlay; Adele Woodhouse; Age, but Not Amyloidosis, Induced Changes in Global Levels of Histone Modifications in Susceptible and Disease-Resistant Neurons in Alzheimer’s Disease Model Mice. Frontiers in Aging Neuroscience 2019, 11, 68, 10.3389/fnagi.2019.00068.
- Hernán Guillermo Hernández; Adrián Gabriel Sandoval-Hernández; Pablo Garrido-Gil; José Luis Labandeira-Garcia; María Victoria Zelaya; Gustavo F Bayon; Agustín F Fernández; Mario F Fraga; Gonzalo Arboleda; Humberto Arboleda; et al. Alzheimer's disease DNA methylome of pyramidal layers in frontal cortex: laser-assisted microdissection study. Epigenomics 2018, 10, 1365-1382, 10.2217/epi-2017-0160.
- Min Young Lee; Junghee Lee; Seung Jae Hyeon; Hyesun Cho; Yu Jin Hwang; Jong-Yeon Shin; Ann C. McKee; Neil W. Kowall; Jong-Il Kim; Thor D. Stein; et al.Daehee HwangHoon Ryu Epigenome signatures landscaped by histone H3K9me3 are associated with the synaptic dysfunction in Alzheimer's disease. Aging Cell 2020, 19, e13153, 10.1111/acel.13153.
- Oliver Kaut; Alfredo Ramirez; Heike Pieper; Ina Schmitt; Frank Jessen; Ullrich Wüllner; DNA Methylation of the TNF-α Promoter Region in Peripheral Blood Monocytes and the Cortex of Human Alzheimer's Disease Patients. Dementia and Geriatric Cognitive Disorders 2014, 38, 10-15, 10.1159/000357126.
- Alison Bernstein; Yunting Lin; R. Craig Street; Li Lin; Qing Dai; Li Yu; Han Bao; Marla Gearing; James J. Lah; Peter T. Nelson; et al.Chuan HeAllan I. LeveyJennifer G. MulléRanhui DuanPeng Jin 5-Hydroxymethylation-associated epigenetic modifiers of Alzheimer’s disease modulate Tau-induced neurotoxicity. Human Molecular Genetics 2016, 25, 2437-2450, 10.1093/hmg/ddw109.
- Zhan Gao; Hong‑Juan Fu; Li‑Bo Zhao; Zhuo‑Yan Sun; Yu‑Fei Yang; Hong‑Yan Zhu; Aberrant DNA methylation associated with Alzheimer's disease in the superior temporal gyrus. Experimental and Therapeutic Medicine 2017, 15, 103-108, 10.3892/etm.2017.5394.
- Aparna Vasanthakumar; Justin W. Davis; Kenneth Idler; Jeffrey F. Waring; Elizabeth Asque; Bridget Riley-Gillis; Shaun Grosskurth; Gyan Srivastava; Sungeun Kim; Kwangsik Nho; et al.Kelly N. H. NudelmanKelley FaberYu SunTatiana M. ForoudKarol EstradaLiana G. ApostolovaQingqin S. LiAndrew J. SaykinFor The Alzheimer’S Disease Neuroimaging Initiative (Adni) Harnessing peripheral DNA methylation differences in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) to reveal novel biomarkers of disease. Clinical Epigenetics 2020, 12, 1-11, 10.1186/s13148-020-00864-y.
- Andrea Stoccoro; Pierpaola Tannorella; Lucia Migliore; Fabio Coppedè; Polymorphisms of genes required for methionine synthesis and DNA methylation influence mitochondrial DNA methylation. Epigenomics 2020, 12, 1003-1012, 10.2217/epi-2020-0041.
- Peter Fransquet; Paul Lacaze; Richard Saffery; James Phung; Emily Parker; Raj Shah; Anne Murray; Robyn L. Woods; Joanne Ryan; Blood DNA methylation signatures to detect dementia prior to overt clinical symptoms. Alzheimer's & Dementia: Diagnosis, Assessment & Disease Monitoring 2020, 12, e12056, 10.1002/dad2.12056.
- J. Mendiola-Precoma; L. C. Berumen; K. Padilla; G. Garcia-Alcocer; Therapies for Prevention and Treatment of Alzheimer’s Disease. BioMed Research International 2016, 2016, 1-17, 10.1155/2016/2589276.
- Wang-Xia Wang; Bernard W. Rajeev; Arnold J. Stromberg; N. Ren; Guiliang Tang; Qingwei Huang; Isidore Rigoutsos; Peter T. Nelson; The Expression of MicroRNA miR-107 Decreases Early in Alzheimer's Disease and May Accelerate Disease Progression through Regulation of -Site Amyloid Precursor Protein-Cleaving Enzyme 1. The Journal of Neuroscience 2008, 28, 1213-1223, 10.1523/jneurosci.5065-07.2008.
- Athanasios Zovoilis; Hope Y Agbemenyah; Roberto Carlos Agís-Balboa; Roman Stilling; Dieter Edbauer; Pooja Rao; Laurent Farinelli; Ivana Delalle; Andrea Schmitt; Peter Falkai; et al.Sanaz Bahari-JavanSusanne BurkhardtFarahnaz SananbenesiAndre Fischer microRNA-34c is a novel target to treat dementias. The EMBO Journal 2011, 30, 4299-4308, 10.1038/emboj.2011.327.
- Lucía Chávez-Gutiérrez; Leen Bammens; Iryna Benilova; Annelies Vandersteen; Manasi Benurwar; Marianne Borgers; Sam Lismont; Lujia Zhou; Simon Van Cleynenbreugel; Hermann Esselmann; et al.Jens WiltfangLutgarde SerneelsEric KarranHarrie GijsenJoost SchymkowitzFrederic RousseauKerensa BroersenBart De Strooper The mechanism of γ-Secretase dysfunction in familial Alzheimer disease. The EMBO Journal 2012, 31, 2261-2274, 10.1038/emboj.2012.79.
- Patricia R. Manzine; Silvia Pelucchi; Maria A. Horst; Francisco A.C. Vale; Sofia C.I. Pavarini; Matteo Audano; Nico Mitro; Monica Di Luca; Elena Marcello; Márcia R. Cominetti; et al. microRNA 221 Targets ADAM10 mRNA and is Downregulated in Alzheimer’s Disease. Journal of Alzheimer's Disease 2017, 61, 113-123, 10.3233/JAD-170592.
- Michael T Heneka; Monica J Carson; Joseph El Khoury; Gary E Landreth; Frederic Brosseron; Douglas L Feinstein; Andreas H Jacobs; Tony Wyss-Coray; Javier Vitorica; Richard M Ransohoff; et al.Karl HerrupSally A. FrautschyBente FinsenGuy C BrownAlexei VerkhratskyKoji YamanakaJari KoistinahoEicke LatzAnnett HalleGabor C PetzoldTerrence TownDave MorganMari L ShinoharaV Hugh PerryClive HolmesNicolas G BazanDavid J BrooksStephane HunotBertrand JosephNikolaus DeigendeschOlga GaraschukErik BoddekeCharles A DinarelloJohn C BreitnerGreg M ColeDouglas T GolenbockMarkus P Kummer Neuroinflammation in Alzheimer's disease. The Lancet Neurology 2015, 14, 388-405, 10.1016/s1474-4422(15)70016-5.
- Bing Bai; U1 snRNP Alteration and Neuronal Cell Cycle Reentry in Alzheimer Disease. Frontiers in Aging Neuroscience 2018, 10, 75, 10.3389/fnagi.2018.00075.
- Saeedeh Hosseinian; Ehsan Arefian; Hassan Rakhsh-Khorshid; Mehdi Eivani; Ameneh Rezayof; Hamid Pezeshk; Sayed-Amir Marashi; A meta-analysis of gene expression data highlights synaptic dysfunction in the hippocampus of brains with Alzheimer’s disease. Scientific Reports 2020, 10, 8384, 10.1038/s41598-020-64452-z.
- Schwartz, M.; Arad, M.; Ben-Yehuda, H; Potential immunotherapy for Alzheimer disease and age-related dementia. Dialogues in Clinical Neuroscience 2019, 21, 21-25, 10.31887/dcns.2019.21.1/mschwartz.
- Kuti Baruch; Neta Rosenzweig; Alexander Kertser; Aleksandra Deczkowska; Alaa Mohammad Sharif; Amit Spinrad; Afroditi Tsitsou-Kampeli; Ayelet Sarel; Liora Cahalon; Michal Schwartz; et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nature Communications 2015, 6, 7967, 10.1038/ncomms8967.
- Nicole K. Rogers; Cesar Romero; Carol D. SanMartín; Daniela P. Ponce; Felipe Salech; Mercedes N. López; Alejandra Gleisner; Fabián Tempio; María I. Behrens; Inverse Relationship Between Alzheimer’s Disease and Cancer: How Immune Checkpoints Might Explain the Mechanisms Underlying Age-Related Diseases. Journal of Alzheimer's Disease 2020, 73, 443-454, 10.3233/JAD-190839.
- Juliane Obst; R. Mancuso; E. Simon; D. Gomez-Nicola; PD-1 deficiency is not sufficient to induce myeloid mobilization to the brain or alter the inflammatory profile during chronic neurodegeneration. Brain, Behavior, and Immunity 2018, 73, 708-716, 10.1016/j.bbi.2018.08.006.
- Neta Rosenzweig; Raz Dvir-Szternfeld; Afroditi Tsitsou-Kampeli; Hadas Keren-Shaul; Hila Ben-Yehuda; Pierre Weill-Raynal; Liora Cahalon; Alex Kertser; Kuti Baruch; Ido Amit; et al.Assaf WeinerMichal Schwartz PD-1/PD-L1 checkpoint blockade harnesses monocyte-derived macrophages to combat cognitive impairment in a tauopathy mouse model. Nature Communications 2019, 10, 1-15, 10.1038/s41467-019-08352-5.
- Kangling Zhang; Matthew Schrag; Andrew Crofton; Rishi Trivedi; Harry Vinters; Wolff Kirsch; Targeted proteomics for quantification of histone acetylation in Alzheimer's disease. PROTEOMICS 2012, 12, 1261-1268, 10.1002/pmic.201200010.
- Melissa Mahgoub; Lisa M. Monteggia; A role for histone deacetylases in the cellular and behavioral mechanisms underlying learning and memory. Learning & Memory 2014, 21, 564-568, 10.1101/lm.036012.114.
- Jiah Lim; Yoojin Song; Jung-Hee Jang; Chul-Ho Jeong; Sooyeun Lee; Byoungduck Park; Young Ho Seo; Aspirin-inspired acetyl-donating HDACs inhibitors. Archives of Pharmacal Research 2018, 41, 967-976, 10.1007/s12272-018-1045-z.
- Shraddha R. Bang; Shirishkumar D. Ambavade; Priti G. Jagdale; Prafulla P. Adkar; Arun B. Waghmare; Prashant D. Ambavade; Lacosamide reduces HDAC levels in the brain and improves memory: Potential for treatment of Alzheimer's disease. Pharmacology Biochemistry and Behavior 2015, 134, 65-69, 10.1016/j.pbb.2015.04.011.
- Ling Zhang; Cui Liu; Jie Wu; Jing-Jing Tao; Xiao-Long Sui; Zhi-Gang Yao; Yan-Feng Xu; Lan Huang; Hua Zhu; Shu-Li Sheng; et al.Chuan Qin Tubastatin A/ACY-1215 Improves Cognition in Alzheimer's Disease Transgenic Mice. Journal of Alzheimer's Disease 2014, 41, 1193-1205, 10.3233/JAD-140066.
- Janine Arts; Peter King; Ann Mariën; Wim Floren; Ann Beliën; Lut Janssen; Isabelle Pilatte; Bruno Roux; Laurence DeCrane; Ron Gilissen; et al.Ian HicksonVeronique VreysEugene CoxKees BolWillem TalloenIlse GorisLuc AndriesMarc Du JardinMichel JanicotMartin PageKristof Van EmelenPatrick Angibaud JNJ-26481585, a Novel “Second-Generation” Oral Histone Deacetylase Inhibitor, Shows Broad-Spectrum Preclinical Antitumoral Activity. Clinical Cancer Research 2009, 15, 6841-6851, 10.1158/1078-0432.ccr-09-0547.
- Yitshak I. Francis; Mauro Fà; Haider Ashraf; Hong Zhang; Agnieszka Staniszewski; David S. Latchman; Ottavio Arancio; Dysregulation of Histone Acetylation in the APP/PS1 Mouse Model of Alzheimer's Disease. Journal of Alzheimer's Disease 2009, 18, 131-139, 10.3233/JAD-2009-1134.
- Claude-Henry Volmar; Hasib Salah-Uddin; Karolina J. Janczura; Paul Halley; Guerline Lambert; Andrew Wodrich; Sivan Manoah; Nidhi H. Patel; Gregory C. Sartor; Neil Mehta; et al.Nancy T. H. MilesSachi DesseDavid DorciusMichael D. CameronShaun P. BrothersClaes Wahlestedt M344 promotes nonamyloidogenic amyloid precursor protein processing while normalizing Alzheimer’s disease genes and improving memory. Proceedings of the National Academy of Sciences 2017, 114, E9135-E9144, 10.1073/pnas.1707544114.
- Hong Qing; Guiqiong He; Philip T. T. Ly; Christopher J. Fox; Matthias Staufenbiel; Fang Cai; Zhuohua Zhang; Shengcai Wei; Xiulian Sun; Chia-Hsiung Chen; et al.Weihui ZhouKe WangWeihong Song Valproic acid inhibits Aβ production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. Journal of Experimental Medicine 2008, 205, 2781-2789, 10.1084/jem.20081588.
- Ana Ricobaraza; Mar Cuadrado-Tejedor; Sonia Marco; Isabel Pérez-Otaño; Ana García-Osta; Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2010, 22, 1040-1050, 10.1002/hipo.20883.
- Sheng-Jun Fan; Fang-I Huang; Jing-Ping Liou; Chia-Ron Yang; The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death & Disease 2018, 9, 1-14, 10.1038/s41419-018-0688-5.
- Kim N. Green; Joan S. Steffan; Hilda Martínez-Coria; Xuemin Sun; Steven S. Schreiber; Leslie Michels Thompson; Frank M. LaFerla; Nicotinamide Restores Cognition in Alzheimer's Disease Transgenic Mice via a Mechanism Involving Sirtuin Inhibition and Selective Reduction of Thr231-Phosphotau. The Journal of Neuroscience 2008, 28, 11500-11510, 10.1523/jneurosci.3203-08.2008.
- Jinhui Hu; Baijiao An; Tingting Pan; Zhengcunxiao Li; Ling Huang; Xingshu Li; Design, synthesis, and biological evaluation of histone deacetylase inhibitors possessing glutathione peroxidase-like and antioxidant activities against Alzheimer’s disease. Bioorganic & Medicinal Chemistry 2018, 26, 5718-5729, 10.1016/j.bmc.2018.10.022.
- Mar Cuadrado-Tejedor; Carolina Garcia-Barroso; Juan A Sanchez-Arias; Obdulia Rabal; Marta Pérez González; Sara Mederos; Ana Ugarte; Rafael Franco; Victor Segura; Gertrudis Perea; et al.Julen OyarzabalAna Garcia-Osta A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer’s Disease Mice. Neuropsychopharmacology 2016, 42, 524-539, 10.1038/npp.2016.163.
- Mar Cuadrado-Tejedor; Marta Pérez González; Cristina García-Muñoz; Damián Muruzabal; Carolina García-Barroso; Obdulia Rabal; Víctor Segura; Juan A. Sánchez-Arias; Julen Oyarzabal; Ana Garcia-Osta; et al. Taking Advantage of the Selectivity of Histone Deacetylases and Phosphodiesterase Inhibitors to Design Better Therapeutic Strategies to Treat Alzheimer’s Disease. Frontiers in Aging Neuroscience 2019, 11, 149, 10.3389/fnagi.2019.00149.


