Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jarosław Przybyciński | + 2472 word(s) | 2472 | 2021-12-13 04:53:48 | | | |
| 2 | Catherine Yang | Meta information modification | 2472 | 2021-12-23 01:49:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Przybyciński, J. Endothelial Glucocorticoid Receptor in Kidney Diseases. Encyclopedia. Available online: https://encyclopedia.pub/entry/17461 (accessed on 26 July 2026).
Przybyciński J. Endothelial Glucocorticoid Receptor in Kidney Diseases. Encyclopedia. Available at: https://encyclopedia.pub/entry/17461. Accessed July 26, 2026.
Przybyciński, Jarosław. "Endothelial Glucocorticoid Receptor in Kidney Diseases" Encyclopedia, https://encyclopedia.pub/entry/17461 (accessed July 26, 2026).
Przybyciński, J. (2021, December 22). Endothelial Glucocorticoid Receptor in Kidney Diseases. In Encyclopedia. https://encyclopedia.pub/entry/17461
Przybyciński, Jarosław. "Endothelial Glucocorticoid Receptor in Kidney Diseases." Encyclopedia. Web. 22 December, 2021.
Copy Citation
Glucocorticoids, as multifunctional hormones, are widely used in the treatment of various diseases including nephrological disorders. They are known to affect immunological cells, effectively treating many autoimmune and inflammatory processes. Furthermore, there is a growing body of evidence demonstrating the potent role of glucocorticoids in non-immune cells such as podocytes. Moreover, novel data show additional pathways and processes affected by glucocorticoids, such as the Wnt pathway or autophagy. The endothelium is currently considered as a key organ in the regulation of numerous kidney functions such as glomerular filtration, vascular tone and the regulation of inflammation and coagulation.
glucocorticoids
glucocorticoid receptor
kidney
1. Introduction
Glucocorticoids (GCs) are multifunctional hormones affecting the human metabolism, the immunological system, reproduction, circadian rhythm and several other vital functions. This implies the importance of GCs in the treatment of a wide spectrum of diseases such as autoimmune and inflammatory processes and malignancies [1][2]. For many years, they have been a staple in the treatment of a variety of kidney diseases such as glomerulonephritis [3]. Despite this success, they can cause several side effects such as infections or compromised metabolism [4]. In some cases, treatment resistance might occur [4]. Those factors increase interest in GC function and signalling in different cells and tissues to minimise side effects and increase the efficacy of GC curation. On the other hand, novel studies have unravelled the roles of GCs and their receptors in the pathology of different diseases and processes [4]. Furthermore, GCs play a variety of roles, sometimes even opposing ones, in different tissues and organs [5]. Research in recent years brought evidence of multifactorial effect of sex hormones and mineralocorticoids on kidney function and structure, especially in diabetic kidney disease [6][7]. Moreover, other studies revealed a wide-array of functions of nuclear receptors in podocytes [8]. One novel study drew attention to the endothelial glucocorticoid receptor function in diabetic kidney disease [9]. In this review, we will focus on the role of the endothelial glucocorticoid receptor (GR) in the pathogenesis of kidney diseases (Table 1).
Table 1. Patomechanism and potential role of endothelial glucocorticoid receptor in the pathogenesis of particular diseases.
| Endothelial Glucocorticoid Receptor in the Pathogenesis of Particular Diseases | Pathomechanism |
|---|---|
| Hypertension and vascular diseases |
|
| Cardio-vascular diseases |
|
| Diabetic nephropathy |
|
| Chronic kidney disease |
|
In humans, GCs are produced mainly by the adrenal glands under the regulation of the hypothalamic–pituitary–adrenal axis and their secretion depends on various factors such as stress or daily rhythm [2]. There is also evidence of local production of GCs in other organs, mainly immunocompetent organs such as the skin, thymus or intestines, although their function is still uncertain [10].
2. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Arterial Hypertension and Vascular Diseases
Hypertension is a common feature of Cushing syndrome which also affects nearly 20% of patients treated with GCs. The pathogenesis of this event is complex and still uncertain, affecting GC signalling in different tissues. Traditionally, it has been associated with mineralocorticoid function and sodium conservation, taking place mainly in the distal nephron, but novel data suggest that many other mechanisms might be involved in this process. Numerous studies, chiefly in animals, have pointed out the important role of vascular contractility affected by both endothelium and smooth muscle cells. In smooth muscles, GCs increase AT1 receptor concentration and change the flow of Na+ and Ca2+. In the central nervous system, GCs disturb neuronal NO release [11]. Salt-sensitive hypertension occurring in individuals with reduced kidney mass, both congenital and for example after unilateral nephrectomy, might be associated more with the function of GR than MR. A controlled study on rats fed a high-salt diet after unilateral nephrectomy showed that hypertension can be reversed by an MR antagonist but not an aldosterone synthase inhibitor. A significant disbalance of 11β-hydroxysteroid dehydrogenases, favouring the production of active GR in kidney tissue, has also been found. This can be explained by the fact that GCs have similar affinity for both GR and MR and their action in kidney is controlled by 11β-hydroxysteroid dehydrogenase 2 and local inactivation [12]. In so-called aldosterone-sensitive distal nephron, this enzymatic deactivation protects kidney epithelial cells from GC-mediated activation and allows for more precise regulation by mineralocorticoids [13]. A study on MR knockout mice demonstrated that expression of the epithelial Na+ channel and improvement of mineral balance can be mediated solely by GC therapy [14]. Interestingly, research on distal nephron GR knockout mice demonstrated that its function is not required for the development and maintenance of GC-induced hypertension [15]. Furthermore, another study on mice with local, tubular GR knockout showed only a transient effect on sodium handling despite the reduction in the sodium chloride cotransporter (NCC) expression [16]. On the other hand, GCs downregulate and inhibit vasopressin receptor, V2R, in the rat inner medullary collecting duct, causing water and sodium secretion after water overload [17].
GCs can cause various effects in ECs associated with blood pressure regulation. Primarily, they can increase the secretion of ET-1 and AT-2 from ECs, causing vascular smooth muscle contraction [18]. Secondarily, they affect NO action in many stages. GCs directly reduce eNOS transcription through GATA interaction in human cell lines. Furthermore, they increase eNOS mRNA degradation and reduce protein stability. In addition, GCs can lower intracellular Ca2+ mobilisation and tamper with the reaction of cells to such stimulants as ATP. The next potential mechanism reducing NO synthesis could be the inhibition of GTP cyclohydrolase—the enzyme responsible for the production of tetrahydrobiopterin, an eNOS cofactor. In animals and in cell cultures, GCs reduce prostacyclin production via phospholipase A2 and potentially COX-1 inhibition (in foetal cells) [19]. A study on mice demonstrated that endothelial GR knockout animals do not develop hypertension after dexamethasone administration. Although those mice had a higher baseline blood pressure (BP) than matched controls, their BP did not change significantly on administration of early or chronic oral dexamethasone. Endothelial GR knockout animals did not present elevated natriuresis in contrast to the control group. Both groups showed a similar decrease in NO production after dexamethasone treatment, but GR knockout mice had a decreased arteriole contractile response to the drug. Interestingly, vascular reactivity to phenylephrine was similar in both groups. In the case of circadian BP rhythm, while it changed significantly in both groups, leading to higher BP in resting hours, it partly returned to normal after 2–7 days in endothelial GR knockout mice [20]. A different study demonstrated that dexamethasone administration increases ROS production in HUVECs as another factor interfering with NO production [21].
In an animal sepsis model, endothelial GR plays a protective role by mitigating iNOS and eNOS activation and might be seen as a negative regulator of eNOS. Endothelial GR knockout mice had a higher mortality rate after LPS injection due to cardiovascular shock. Moreover, studies on HUVECs have shown that GR knockdown by siRNA augments the inflammatory response by upregulation of the NF-κB pathway [22]. This pathological effect could not be reversed by previous dexamethasone administration. In fact, endothelial GR knockout mice had worse prognosis with additional dexamethasone treatment. These studies indicate the importance of endothelial GR in the pathogenesis of septic shock and BP regulation in animal models [23]. On the other hand, previous studies have demonstrated that dexamethasone can activate eNOS in a non-genomic manner in a mouse model of ischaemia–reperfusion injury. In this model, GCs showed a protective role in reducing vascular inflammation and reducing the myocardial infarct area [24].
At the moment, it is assumed that atherosclerosis originates due to two events: EC damage and vascular remodelling. The consequences are increased endothelial permeability and recruitment of circulating inflammatory cells through ICAM-1 and VCAM-1 expression. This allows the migration of LDL into the vascular wall and further inflammation. Traditionally, an excess of GCs, as observed in Cushing syndrome, is connected with higher cardiovascular risk and development of atherosclerosis but novel studies of both animal models and cell lines bring conflicting results. Despite the known role of GCs in the downregulation of EC adhesion molecules and inhibition of IL-6, IL-8 and CCL which should be beneficial, some animal models show pro-atherogenic effects [18].
Endothelial GR is important in the pathophysiology of atherosclerosis. Studies using endothelial GR knockout mice fed with a high-fat diet showed greater atherosclerosis in comparison to wild genotype mice. It is postulated that the action of endothelial GR attenuates atherosclerosis and vascular inflammation [25]. Due to the multifactorial pathophysiology of atherosclerosis, the effect might be dependent on the different cells involved and other aspects [18].
3. Role of Endothelial Glucocorticoid Receptor in the Pathogenesis of Glomerulopathies
For many years, GCs have been used as a cornerstone in the therapy of most types of glomerulonephritis, both primary and secondary, and in some, such as minimal change disease, they can be successfully used in monotherapy. In others, such as crescentic glomerulonephritis in vasculitis or severe cases of lupus nephritis, they fail to establish sustainable remission without the addition of other immunosuppressive drugs [3]. These heterogeneous clinical results have generated interest in the precise action of GCs in glomeruli. The therapeutic effect of GCs had been traditionally attributed to the mitigation of inflammation directly in immune cells. Novel studies unravel a more complex relationship. For example, GCs protect podocytes through stabilisation of the slit diaphragm and cytoskeleton and preserve podocyte differentiation after injury [26]. Moreover, mice with GR knockout in podocytes are more vulnerable to kidney injury by various factors and show worse proteinuria and podocyte foot process effacement. This has been attributed to defective cytoskeleton formation and migration after podocyte injury [27]. Interestingly, some studies have revealed a potential negative effect of GR stimulation. One study analysed GR inactivation (selective GR knockout or GR antagonist treatment) in kidney epithelial cells in a mouse model of crescentic glomerulonephritis. It led to a beneficial effect, reduced albuminuria and reduced crescent formation parallel to GC treatment results, but without any major side effects. This was achieved chiefly by inhibition of peripheral epithelial cell proliferation and activating a process associated with crescent formation [28]. The role of endothelial GR in glomerulopathies is less elucidated (Table 2). We already know that normal structure and function of endothelium is crucial for glomerular filtration and protection against thrombosis and inflammation. Based on the data from animals, cell lines and human tissue samples, we know that NF-κB signalling in ECs mediated by ANCA-stimulated neutrophils is a key pathogenetic factor causing ANCA-associated vasculitis [29]. Another important process responsible for appropriate glomerular filtration barrier function is podocyte–endothelium crosstalk (Figure 1). One of the major factors involved in this action is VEGF [30]. Mice overexpressing VEGFR in podocytes develop a nephrotic syndrome similar to minimal change disease observed in humans. It is mediated chiefly by podocyte foot process effacement without significant endothelial damage [31]. It has been previously demonstrated that plasma and urinary VEGF levels are elevated during the onset of nephrotic syndrome in humans and decline after GC treatment [32]. On the other hand, VEGF depletion can cause endothelial injury and proteinuria in a process called endotheliosis. It is assumed as a main pathophysiological lesion in pre-eclampsia or anti-VEGF cancer treatment complications [30][33][34].
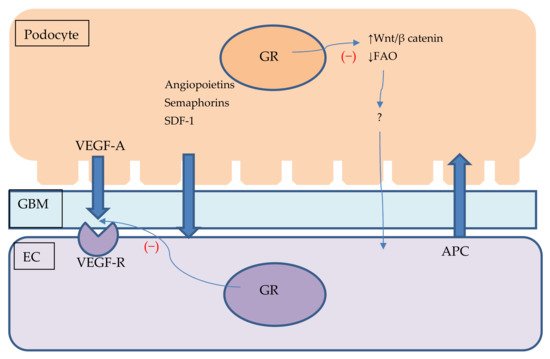
Figure 1. Podocyte–endothelial crosstalk and its potential connection with GR signalling. Figure shows podocyte and endothelial cell with glomerular base membrane between them. Podocytes produce VEGF, angiopoietins, semaphorins and SDF-1 which are secreted and influence endothelial cells homeostasis. ECs might secrete APC which protects podocytes against apoptosis. GR stimulation might impair VEGF signalling and influence podocyte Wnt/β catenin pathway and promote FAO. Lack of GR stimulation in podocytes might damage ECs through unknown mediator. APC—activated protein C, EC—endothelial cell, FAO—fatty acid oxidation, GBM—glomerular base membrane, SDF-1—Stromal cell-derived factor 1 VEGF—vascular endothelial growth factor, VEGF-R—vascular endothelial growth factor receptor, Wnt—Wnt signalling pathway.
Table 2. Potential links between glomerulopathies and abnormal GR signalling.
| Glomerulonephritis | Pathogenesis of Selected Glomerulonephritis |
|---|---|
| Focal segmental glomerulosclerosis (FSGS) |
|
| ANCA-associated vasculitis |
|
| Minimal change disease |
|
| Lupus nephritis |
|
Glomerular ECs compared with other ECs produce more fibrinolytic factors and have a different metabolic profile, leading to their vulnerability to oxidative stress and membrane lipid peroxidation [35]. Damage to ECs is primarily responsible for the pathogenesis of lupus nephritis, vasculitis, TMA and antibody-mediated rejection of kidney transplant [36][37][38].
Previous studies have demonstrated a potent anti-apoptotic effect of GCs in cultured bovine ECs. Exposure of cells to TNF-α or LPS causes apoptosis and this effect is inhibited by the addition of dexamethasone. There is a time window up to 18 h after cell insult when dexamethasone application shows a protective outcome. The nature of this reaction is still unclear. It has been clearly attributed to GR action and processes upstream of caspase activation, mainly changes in the composition of bcl-2-related proteins. On the other hand, the anti-apoptotic effect could be only partly linked with the early stages of apoptosis activation and mitochondrial permeability transition. Similar results of an anti-apoptotic effect of dexamethasone have been observed in an EC line exposed to fluvastatin [39][40][41].
Some types of glomerulonephritis, for example, those associated with vasculitis, cannot be treated effectively with GC monotherapy. An insight into this phenomenon has been given by analysing the effect of GCs in different cells. A study on cell cultures comparing the reaction to GCs in monocytes and ECs in standard and inflammatory milieus showed a contrasting result. In monocytes, GCs suppressed pro-inflammatory genes such as TNFSF10, IL1B and CCL5 and induced anti-inflammatory genes such as IL1R2, DUSP1, FPR1 and FKBP5. Interestingly, GCs failed to present this effect in HUVEC cultures. Further analysis and gene profiling revealed that while GCs suppressed 22% to 28% of immune response-associated genes in monocytes, they were able to suppress only 2% of those genes in HUVECs. This difference in GC action could not be explained by different expression of GR or its impaired translocation but might be associated with decreased induction of SAP30—a subunit of the Sin3A–HDAC corepressor complex [42]. Under other conditions, previous studies have shown that GCs can decrease activation of the MAPK signalling pathway, providing an anti-inflammatory effect in human microvascular ECs acquired from lungs. These results have not been tested in ECs acquired from kidneys [43]. In a different study, GCs inhibited IL-6 production but failed to interact with VCAM-1 induction in HUVECs under inflammatory conditions. This could be explained by the different coactivators necessary for GR–NF-κB interaction with IL-6 and VCAM-1 promotors [44].
References
- Reichardt, S.D.; Amouret, A.; Muzzi, C.; Vettorazzi, S.; Tuckermann, J.P.; Lühder, F.; Reichardt, H.M. The Role of Glucocorticoids in Inflammatory Diseases. Cells 2021, 10, 2921.
- Noureddine, L.M.; Trédan, O.; Hussein, N.; Badran, B.; Le Romancer, M.; Poulard, C. Glucocorticoid Receptor: A Multifaceted Actor in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 4446.
- Ponticelli, C.; Locatelli, F. Glucocorticoids in the Treatment of Glomerular Diseases: Pitfalls and Pearls. Clin. J. Am. Soc. Nephrol. 2018, 13, 815–822.
- Timmermans, S.; Souffriau, J.; Libert, C. A General Introduction to Glucocorticoid Biology. Front. Immunol. 2019, 10, 1545.
- Whirledge, S.; DeFranco, D.B. Glucocorticoid Signaling in Health and Disease: Insights From Tissue-Specific GR Knockout Mice. Endocrinology 2018, 159, 46–64.
- Giandalia, A.; Giuffrida, A.E.; Gembillo, G.; Cucinotta, D.; Squadrito, G.; Santoro, D.; Russo, G.T. Gender Differences in Diabetic Kidney Disease: Focus on Hormonal, Genetic and Clinical Factors. Int. J. Mol. Sci. 2021, 22, 5808.
- Vodošek Hojs, N.; Bevc, S.; Ekart, R.; Piko, N.; Petreski, T.; Hojs, R. Mineralocorticoid Receptor Antagonists in Diabetic Kidney Disease. Pharmaceuticals 2021, 14, 561.
- Agrawal, S.; He, J.C.; Tharaux, P.L. Nuclear receptors in podocyte biology and glomerular disease. Nat. Rev. Nephrol. 2021, 17, 185–204.
- Srivastava, S.P.; Zhou, H.; Setia, O.; Dardik, A.; Fernandez-Hernando, C.; Goodwin, J. Podocyte Glucocorticoid Receptors Are Essential for Glomerular Endothelial Cell Homeostasis in Diabetes Mellitus. J. Am. Heart Assoc. 2021, 10, e019437.
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-adrenal glucocorticoids and mineralocorticoids: Evidence for local synthesis, regulation, and function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24.
- Goodwin, J.E.; Geller, D.S. Glucocorticoid-induced hypertension. Pediatr. Nephrol. 2012, 27, 1059–1066.
- Huesler, C.; Lauterburg, M.; Frey, B.M.; Frey, F.J. Evidence for glucocorticoid-mediated hypertension after uninephrectomy. Physiol. Rep. 2013, 1, e00101.
- Liu, B.; Zhang, T.N.; Knight, J.K.; Goodwin, J.E. The Glucocorticoid Receptor in Cardiovascular Health and Disease. Cells 2019, 8, 1227.
- Schulz-Baldes, A.; Berger, S.; Grahammer, F.; Warth, R.; Goldschmidt, I.; Peters, J.; Schütz, G.; Greger, R.; Bleich, M. Induction of the epithelial Na+ channel via glucocorticoids in mineralocorticoid receptor knockout mice. Pflug. Arch. 2001, 443, 297–305.
- Goodwin, J.E.; Zhang, J.; Velazquez, H.; Geller, D.S. The glucocorticoid receptor in the distal nephron is not necessary for the development or maintenance of dexamethasone-induced hypertension. Biochem. Biophys. Res. Commun. 2010, 394, 266–271.
- Canonica, J.; Frateschi, S.; Boscardin, E.; Ebering, A.; Sergi, C.; Jäger, Y.; Peyrollaz, T.; Mérillat, A.M.; Maillard, M.; Klusonova, P.; et al. Lack of Renal Tubular Glucocorticoid Receptor Decreases the Thiazide-Sensitive Na. Front. Physiol. 2019, 10, 989.
- Zhu, X.; Huang, Y.; Li, S.; Ge, N.; Li, T.; Wang, Y.; Liu, K.; Liu, C. Glucocorticoids Reverse Diluted Hyponatremia Through Inhibiting Arginine Vasopressin Pathway in Heart Failure Rats. J. Am. Heart Assoc. 2020, 9, e014950.
- MacLeod, C.; Hadoke, P.W.F.; Nixon, M. Glucocorticoids: Fuelling the Fire of Atherosclerosis or Therapeutic Extinguishers? Int. J. Mol. Sci. 2021, 22, 7622.
- Yang, S.; Zhang, L. Glucocorticoids and vascular reactivity. Curr. Vasc. Pharmacol. 2004, 2, 1–12.
- Goodwin, J.E.; Zhang, J.; Gonzalez, D.; Albinsson, S.; Geller, D.S. Knockout of the vascular endothelial glucocorticoid receptor abrogates dexamethasone-induced hypertension. J. Hypertens. 2011, 29, 1347–1356.
- Iuchi, T.; Akaike, M.; Mitsui, T.; Ohshima, Y.; Shintani, Y.; Azuma, H.; Matsumoto, T. Glucocorticoid excess induces superoxide production in vascular endothelial cells and elicits vascular endothelial dysfunction. Circ. Res. 2003, 92, 81–87.
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Sessa, W.C. Endothelial glucocorticoid receptor is required for protection against sepsis. Proc. Natl. Acad. Sci. USA 2013, 110, 306–311.
- Goodwin, J.E.; Feng, Y.; Velazquez, H.; Zhou, H.; Sessa, W.C. Loss of the endothelial glucocorticoid receptor prevents the therapeutic protection afforded by dexamethasone after LPS. PLoS ONE 2014, 9, e108126.
- Hafezi-Moghadam, A.; Simoncini, T.; Yang, Z.; Limbourg, F.P.; Plumier, J.C.; Rebsamen, M.C.; Hsieh, C.M.; Chui, D.S.; Thomas, K.L.; Prorock, A.J.; et al. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat. Med. 2002, 8, 473–479.
- Goodwin, J.E.; Zhang, X.; Rotllan, N.; Feng, Y.; Zhou, H.; Fernández-Hernando, C.; Yu, J.; Sessa, W.C. Endothelial glucocorticoid receptor suppresses atherogenesis--brief report. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 779–782.
- Goodwin, J.E. Role of the glucocorticoid receptor in glomerular disease. Am. J. Physiol. Renal Physiol. 2019, 317, F133–F136.
- Zhou, H.; Tian, X.; Tufro, A.; Moeckel, G.; Ishibe, S.; Goodwin, J. Loss of the podocyte glucocorticoid receptor exacerbates proteinuria after injury. Sci. Rep. 2017, 7, 9833.
- Kuppe, C.; van Roeyen, C.; Leuchtle, K.; Kabgani, N.; Vogt, M.; Van Zandvoort, M.; Smeets, B.; Floege, J.; Gröne, H.J.; Moeller, M.J. Investigations of Glucocorticoid Action in GN. J. Am. Soc. Nephrol. 2017, 28, 1408–1420.
- Choi, M.; Schreiber, A.; Eulenberg-Gustavus, C.; Scheidereit, C.; Kamps, J.; Kettritz, R. Endothelial NF-κB Blockade Abrogates ANCA-Induced GN. J. Am. Soc. Nephrol. 2017, 28, 3191–3204.
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461.
- Veron, D.; Reidy, K.; Marlier, A.; Bertuccio, C.; Villegas, G.; Jimenez, J.; Kashgarian, M.; Tufro, A. Induction of podocyte VEGF164 overexpression at different stages of development causes congenital nephrosis or steroid-resistant nephrotic syndrome. Am. J. Pathol. 2010, 177, 2225–2233.
- Wasilewska, A.; Zoch-Zwierz, W. Glucocorticoid receptor and vascular endothelial growth factor in nephrotic syndrome. Acta Paediatr. 2006, 95, 587–593.
- Ballermann, B.J. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007, 106, 19–25.
- Eremina, V.; Jefferson, J.A.; Kowalewska, J.; Hochster, H.; Haas, M.; Weisstuch, J.; Richardson, C.; Kopp, J.B.; Kabir, M.G.; Backx, P.H.; et al. VEGF inhibition and renal thrombotic microangiopathy. N. Engl. J. Med. 2008, 358, 1129–1136.
- Betzen, C.; Plotnicki, K.; Fathalizadeh, F.; Pappan, K.; Fleming, T.; Bielaszewska, M.; Karch, H.; Tönshoff, B.; Rafat, N. Shiga Toxin 2a-Induced Endothelial Injury in Hemolytic Uremic Syndrome: A Metabolomic Analysis. J. Infect. Dis. 2016, 213, 1031–1040.
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108.
- Ding, Y.; Tan, Y.; Qu, Z.; Yu, F. Renal microvascular lesions in lupus nephritis. Ren. Fail. 2020, 42, 19–29.
- Amaral, L.M.; Wallace, K.; Owens, M.; LaMarca, B. Pathophysiology and Current Clinical Management of Preeclampsia. Curr. Hypertens. Rep. 2017, 19, 61.
- Messmer, U.K.; Winkel, G.; Briner, V.A.; Pfeilschifter, J. Glucocorticoids potently block tumour necrosis factor-alpha- and lipopolysaccharide-induced apoptotic cell death in bovine glomerular endothelial cells upstream of caspase 3 activation. Br. J. Pharmacol. 1999, 127, 1633–1640.
- Messmer, U.K.; Winkel, G.; Briner, V.A.; Pfeilschifter, J. Suppression of apoptosis by glucocorticoids in glomerular endothelial cells: Effects on proapoptotic pathways. Br. J. Pharmacol. 2000, 129, 1673–1683.
- Newton, C.J.; Ran, G.; Xie, Y.X.; Bilko, D.; Burgoyne, C.H.; Adams, I.; Abidia, A.; McCollum, P.T.; Atkin, S.L. Statin-induced apoptosis of vascular endothelial cells is blocked by dexamethasone. J. Endocrinol. 2002, 174, 7–16.
- Koenen, P.; Barczyk, K.; Wolf, M.; Roth, J.; Viemann, D. Endothelial cells present an innate resistance to glucocorticoid treatment: Implications for therapy of primary vasculitis. Ann. Rheum. Dis. 2012, 71, 729–736.
- Pelaia, G.; Cuda, G.; Vatrella, A.; Grembiale, R.D.; De Sarro, G.; Maselli, R.; Costanzo, F.S.; Avvedimento, V.E.; Rotiroti, D.; Marsico, S.A. Effects of glucocorticoids on activation of c-jun N-terminal, extracellular signal-regulated, and p38 MAP kinases in human pulmonary endothelial cells. Biochem. Pharmacol. 2001, 62, 1719–1724.
- Xu, X.; Otsuki, M.; Saito, H.; Sumitani, S.; Yamamoto, H.; Asanuma, N.; Kouhara, H.; Kasayama, S. PPARalpha and GR differentially down-regulate the expression of nuclear factor-kappaB-responsive genes in vascular endothelial cells. Endocrinology 2001, 142, 3332–3339.
- Gembillo, G.; Siligato, R.; Amatruda, M.; Conti, G.; Santoro, D. Vitamin D and Glomerulonephritis. Medicina 2021, 57, 186.
More
Information
Subjects:
Urology & Nephrology; Cell Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
665
Revisions:
2 times
(View History)
Update Date:
23 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No