The identification and characterisation of genomic changes (variants) that can lead to human diseases is one of the central aims of biomedical research. In order to take on this challenging task, Genome-Wide Association Studies (GWAS) were proposed as a statistical method that could be used to identify the genomic variants that are associated with complex traits or diseases. GWAS do not require any previous biological knowledge on the analyzed trait, as they allow the simultaneous interrogation of millions of variants genome-wide. As a result, GWAS have been largely used, substantially contributing to the generation of catalogues of genetic variants that have an impact on specific diseases. GWAS results constitute nowadays the basis of Personalised Medicine, where diagnoses and treatment protocols are selected according to each patient’s profile.
1. Introduction
Complex traits, such as height, blood pressure, or some types of diseases, arise from the combination of multiple environmental and genetic factors (see
Box 1 for definitions of fundamental concepts). In these, each of the involved genetic variants is expected to only make a marginal contribution to the whole phenotype, each explaining <1% and often <0.5%, of phenotypic variability
[1][2][3]. Consequently, hundreds or even thousands of loci are likely to be involved for each trait
[4][5][6]. Complex diseases, such as diabetes
[7], asthma
[8], cardiovascular diseases
[9], or Alzheimer’s disease
[10], tend to appear late in life and strongly affect the quality of life of millions of individuals around the world, exerting a large economic and social pressure on developed global healthcare systems. For instance, diabetes incurred in an estimated cost of USD 327 billion in 2017 in the United States alone, a value that increased 26% with respect to 2012
[11]. To help alleviate this burden, a long-standing goal of biomedicine has been to gain a better understanding of the molecular mechanisms and the genetic architecture behind these diseases, enabling better prognosis, prevention, and treatment protocols.
In addition to the multifactorial architecture of complex traits, covariate effects, population substructure, or disease heterogeneity
[12] make the identification of the underlying causal genomic variants a statistical, mathematical, and computational challenge. The recent increase in sample sizes and the improvement of statistical frames have helped increase sensibility but have also imposed computational and methodological burdens that are becoming the bottleneck of these types of analyses. This increasing complexity has forced many studies to reduce their overall scope, which they may accomplish by excluding the analysis of the X chromosome or by restricting the analysis of the additive model, disregarding all other inheritance models that should be considered. This substantially limits the chances of identifying novel genetic markers that are associated with disease, as we recently demonstrated
[13][14].
2. Preliminary Genome Biology Concepts
The human genome is considerably variable. Two human beings differ in 4.1–5 million genomic sites on average, for a total of around 20 million bases (~0.6% of the total genome)
[15]. This genetic variability determines not only the differences in physical appearance, such as height or eye colour, but also the predisposition of an idividual to develop diseases.
Distinguishing the genetic variants that are responsible of normal human variability from those affecting disease risk is thus fundamental to predict, diagnose, and possibly treat diseases, contributing to personalised medicine efforts. In this scenario, GWAS represents a resourceful strategy that can be used to identify variants that are associated with complex diseases. Despite substantial advancements, this remains a challenging task: in complex diseases, the contribution of each of the genetic variants to the final phenotype has been proven to be low and to come later in life, which is in contrast to rare diseases, where variants usually have a much stronger effect in the individual and may already be present during early developmental stages
[1][14].
In general terms, each individual inherits this variability through parental germ cells. For example, when the genomic variation consists of a change at a single nucleotide position, it is called a Single Nucleotide Variant (SNV), but larger, structural variants (e.g., duplications, deletions) that have the potential of affecting up to millions of nucleotides also exist (see
Box 2 for definitions of genomic concepts). As a result of the meiosis process, any genomic position (loci) is thus present in two copies (alleles). The set of alleles in a single homologous chromosome is defined as a haplotype, and the combination of all alleles identifies the individual’s genotype. The study of these genotypes in regard to their relationship with diseases is one of the central aims of biomedicine. It allows us to generate comprehensive genetic maps for each disease and to use them to easily screen, for example, newborns and to be able to predict the disease risk for that newborn and to plan preventive protocols.
Most genomic variants are biallelic, meaning that only two different alleles (generally named
A and
B) exist in the population. In this scenario and considering that all individuals have two copies of the genome, at any given variable locus (position), an individual displays one of three possible genotypes:
AA,
AB, or
BB. When compared to the human reference genome
[16], the allele matching the reference (e.g.,
A) is termed the reference allele, while the other (e.g.,
B) is termed the alternate allele. Consequently, the three possible genotypes are labelled as the homozygous reference (
hom. ref. or
AA), the homozygous alternate (
hom. alt. or
BB), or heterozygous (
het. or
AB).
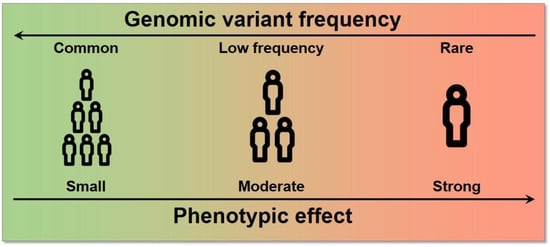
Each of these genetic variants, which likely arose from single different individuals, are spread and fixed within the population over long periods of time and follow evolutionary rules based on the harm or benefit that each variation provides to the individual. As a consequence of this process, variants have different frequencies within each population, as they are carried by different proportions of individuals. Variants with frequencies
are defined as common, while variants with frequencies
or
are defined as low-frequency and rare, respectively. SNVs with a frequency of >1% in the population are typically called Single Nucleotide Polymorphisms (SNPs). Since complex diseases are common, originally, only common variants were considered to be implicated (common disease-common variant hypothesis); the possibility of extending GWAS even to low-frequency and rare variants has shown, however, that variants across the entire frequency spectrum are likely to be involved
[3]. The effect size, which is the contribution of these variants to the phenotype, is generally measured by an odds ratio (the odds of having the disease with the variant divided by the odds of having the disease without it) for a binary trait. Typically, an inverse relationship exists between the frequency of a variant and its effect on diseases: high-impact variants are normally found at lower frequencies because of a stronger negative selection pressure (
Figure 1)
[17].
Figure 1. Relationship between allele frequency and effect size. High effect variants tend to have a lower frequency in the population and vice versa.
Finally, it is worth noting that even though ~50% of the genome is inherited from each parent, the nucleotides in a chromosome are not inherited independently. Instead, the genomic material is exchanged in large, linked fragments, that are delimited by recombination hotspots, which are genomic regions that are more prone to recombination. As a result, these large genomic fragments contain multiple alleles that are inherited as a whole from the same parent; these alleles are said to be in linkage disequilibrium (LD).
3. Genome Wide Association Studies (GWAS)
Definition
In order to take on this challenging task, GWAS was proposed as a statistical method that could be used to identify the genomic variants that are associated with complex traits or diseases. Specifically, GWAS are statistical analyses that aim to find the associations between genomic variability and a particular trait or disease
[17]. Previous studies have required each functional hypothesis to be specifically tested in the context of a disease. In contrast, GWAS allow for the exploration of the genetic architecture of diseases at the genome-wide level, without the need of prior hypotheses beyond the existence of a genetic component behind the disease.
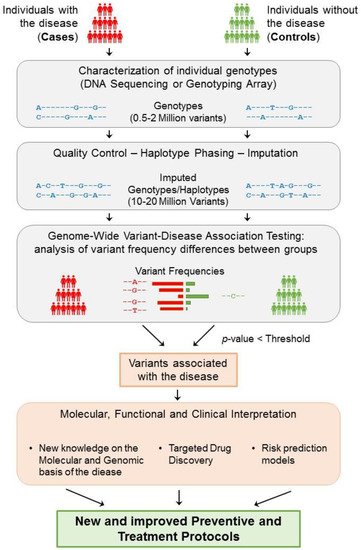
These studies collect genotypes and phenotypes of a large number of participants, generally in the order of tens of thousands, or even millions. To study a complex disease (binary trait), participants are separated into cases (affected) and controls (non-affected) (
Figure 2). Then, a prior characterisation of the variation landscape is needed for each of the participating individuals, i.e., the genotypes and haplotypes, which are inferred from the lists of variants that have been identified within each participant. Whereas whole-genome sequencing currently provides the most complete map of genomic variation for an individual, it is still a very expensive and time-consuming assay, especially when considering the large number of participants within these types of studies. Instead, GWAS typically use DNA hybridisation microarray technologies, a more affordable alternative. DNA microarrays, however, are designed to interrogate only a limited set of pre-selected genomic variants (generally between 500 k and 2 M)
[18]. These variants are chosen to be common across the population, so that many of the individuals can carry them, and are also chosen considering LD blocks, so that only a single variant in each block is typically probed.
Figure 2. General strategy underlying GWAS. The study of a complex disease through GWAS starts with the selection of a large group of individuals that can be segregated into cases (affected) and controls (non-affected). Then, each individual genotype is characterized using DNA sequencing techniques or genotyping arrays, obtaining the genotyping information of 0.5–2 million variants from each individual. After ensuring the quality of these data, phasing and imputation techniques are usually applied to increase the number of variants that can be tested to 10–20 million. Each resulting genomic variant is then independently tested to find significant differences in the genotype frequencies between the two groups. Consequently, if a variant is significantly predominant in a group based on an adjusted p-value threshold, then the variant is said to be associated with the disease. Disease-associated variants can then be further analysed to gain insight into their molecular, functional, and clinical implications. As a result of this process, the knowledge obtained from GWAS can help generate and improve the protocols for the better detection, prevention, and treatment of complex diseases.
Then, each genomic variant is independently tested for significant differences in the genotype frequency between the two groups. Thus, if a variant is found to be present significantly more frequently in cases than they are in controls (or vice versa), then that variant is said to be associated with the disease (Figure 2). If the study is sufficiently powered, then a few genomic loci (containing a small number of variants, typically in high LD) will be identified as being significantly associated with the phenotype. For quantitative traits, the individual phenotypes are usually expressed as a continuous variable, and the association is evaluated based on the correlation between the trait and each variant genotype.
Finally, the genomic variants that are significantly associated with a trait or disease (termed “GWAS variants”) provide a list of candidates for further functional analyses to determine in which way they affect the function of the cell and, in the case of disease, ultimately help provide better prevention and treatment protocols.
 +1 credit
+1 credit