 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Melisse Erasmus | + 3256 word(s) | 3256 | 2020-08-18 03:53:48 | | | |
| 2 | Melisse Erasmus | -78 word(s) | 3178 | 2020-08-20 10:48:43 | | | | |
| 3 | Camila Xu | Meta information modification | 3178 | 2020-08-20 11:18:54 | | |
Video Upload Options
LOXL2 is a key enzyme that catalyzes the cross-linking of collagen fibers and forms an integral part in collagen homeostasis. It is thus needed for normal functioning of the myocardium and is pertinent to cardiac remodeling [13]. Dysregulation of its expression is a major driver of muscle stiffness through induced cardiac fibrosis [24,25,43], which reduces cardiac output. In fact, it has been proposed that decreasing excessive collagen cross-linking would reduce myocardial fibrosis and stiffness and thereby improve heart function [44].
1. The Lysyl Oxidase Gene Family
Lysyl oxidase (LOX) and lysyl oxidase-like 1–4 (LOXL1–4) are a family of proteins that play an essential role in collagen and elastin cross-linking [45]. It is now well established that the LOX protein family is linked to fibrosis, as well as the development of other connective tissue disorders, such as elastolysis and Ehlers-Danlos syndrome [46]. Although differentiation between the various isoforms is still unclear, LOX is known to promote collagen and elastin cross-linking by oxidatively deaminating the lysine and hydroxylysine groups within the peptide chains [46]. In addition, LOX is implicated in the transcriptional regulation of transforming growth factor beta (TGF-β) signaling [47]. Increased TGF-β is known to upregulate of alpha smooth muscle actin (α-SMA) leading to excessive cardiac scar formation with enhanced fibrosis [36,48].
TGF-β and α-SMA form an integral part in the fibrotic signaling pathways along with LOX. Although LOX and the LOXL1–4 isoforms are suspected to function similarly due to the conservation of the catalytic domains, the LOX protein family is predominantly expressed in a variety of tissue types with different substrate preferences [26]. It is known that some of the tissue and substrate targets of the different LOX isoforms may overlap, but the exact localizations and functions of these proteins are not yet clearly defined. As illustrated in Figure 1, the copper binding, lysine tyrosylquinone cofactor, cytokine receptor-like and C-terminal regions are catalytic domains that are conserved across the LOX protein family [49–51]. These conserved domains are necessary for LOX activity, and due to these conserved domains and high protein similarity, it is predicted that LOXL 1–4 isoforms perform cross-linking in a similar manner. Any variations in the function of the LOX-family isoforms is likely to occur due to differences in the variable N-terminal regions of each protein.
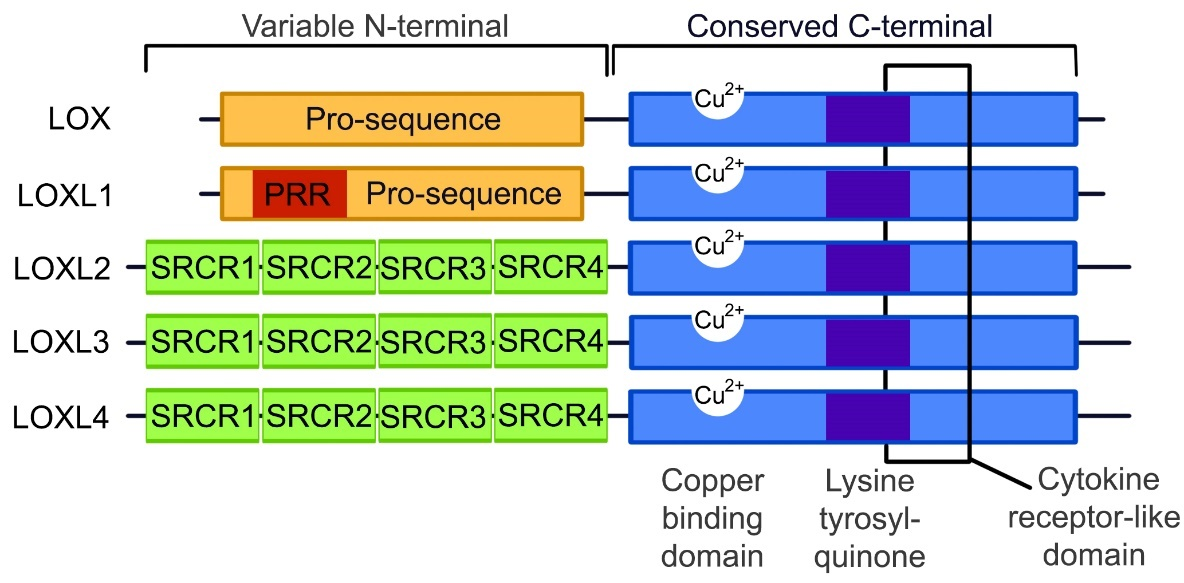
Figure 1. The structure of the LOX and LOXL1–4 proteins. The C-terminal is conserved between all the LOX protein family members, containing a copper binding domain, a lysine tyrosylquinone cofactor residue and a cytokine receptor domain. At the N-terminals, LOX and LOXL1 contain pro-sequences, with LOXL1 containing a proline-rich region (PRR), while LOXL2–4 contain scavenger receptor cysteine-rich domains (SRCR) within the N-terminal. (Image adapted from Wu 2015 [50]).
LOX exists in an inactive form, which is mainly located in the cytosol, and upon activation, it is translocated to the nucleus and endoplasmic reticulum, and secreted into the extracellular space [52]. The inactive form is associated with the endoplasmic reticulum and receives the copper ion cofactor from Menkes’ protein, a copper transporter, to produce the active pro-LOX [26]. Once in the extracellular space, the activated LOX is known to interact with procollagen molecules, and oxidatively deaminates lysine and hydroxylysine residues in the telopeptide portions of the fibers to form lysine aldehyde and hydroxylysine aldehyde, respectively [19]. Once the propeptide fragments of the procollagen molecules are truncated, forming tropocollagen molecules, they self-assemble, and the aldehyde portions form covalent bonds to cross-link the collagen molecules to form fibrils [19]. The mechanisms involved in LOX-dependent cross-linking are depicted in Figure 2, with the process thought to be similar for LOXL1-4.
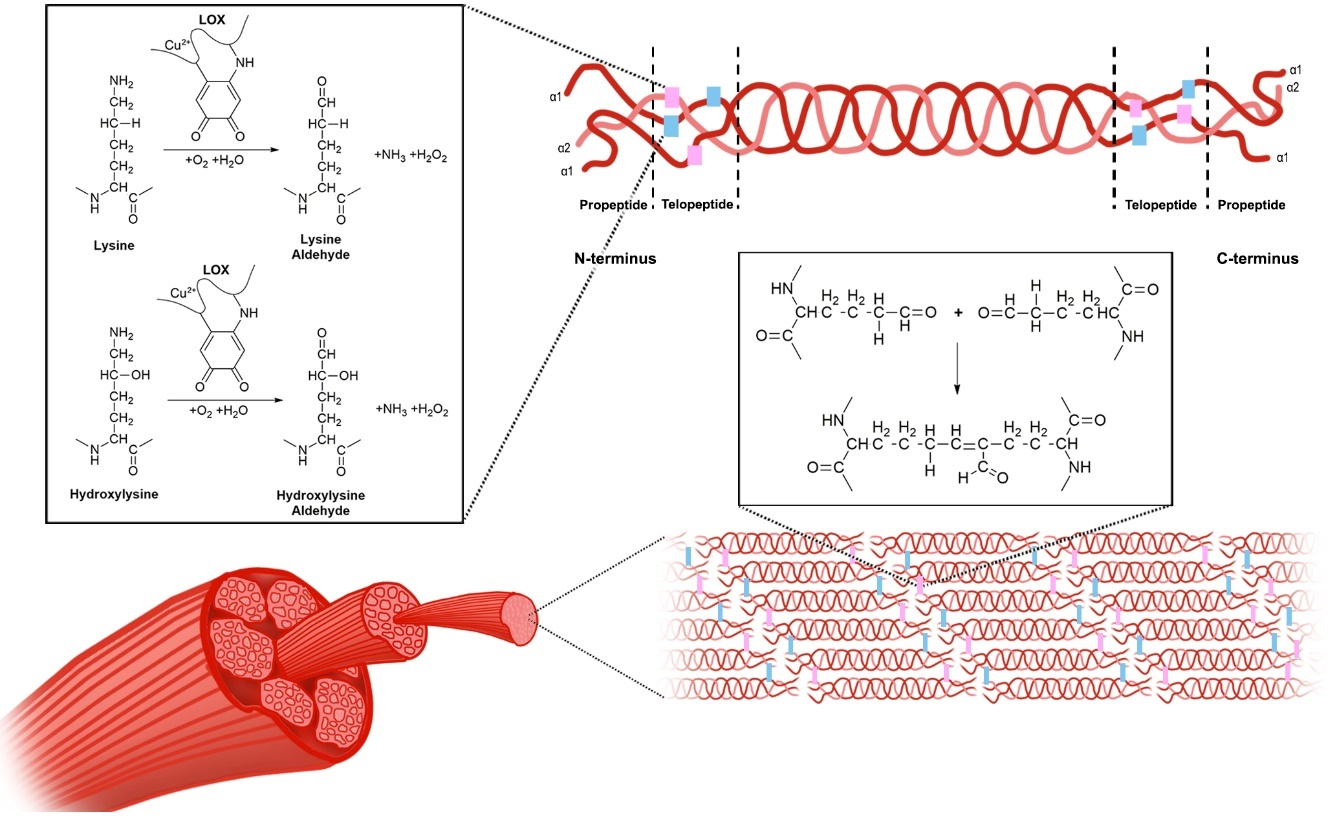
Figure 2. The mechanism of lyslyl oxidase collagen cross-linking. LOX catalyzes the conversion of lysine and hydroxylysine to lysine aldehyde and hydroxylysine aldehyde, respectively. This occurs within the telopeptide region of the procollagen molecules. The propeptide fragments of these molecules are then truncated to form tropocollagen molecules, which self-assemble and form cross-links, thereby forming collagen fibrils.
Apart from collagen and elastin cross-linking, the LOX protein isoforms perform additional functions because of the substrate preference of each enzyme. For example, LOX knock-down studies have shown that it is necessary for the proliferation and differentiation of some cell types, such as osteoblasts [53]. LOX also has the ability to inactivate TGF-β and fibroblast growth factor 2 (FGF-2) by means of oxidation, thus controlling the differentiation of myofibroblasts to fibroblasts [54,55]. Liu et al. (2004) reported that LOXL1 displays substrate preference for elastin and in knockout mouse studies, has been associated with development of abnormal vasculature [56]. In addition, it was shown by Ohmura et al. (2012) that transgenic expression of LOXL1 in mouse hearts induced cardiac hypertrophy [57]. Like LOXL1, Busnadiego et al. (2013) [58] reported that the LOXL4 isoform is involved in vascular remodeling, although it has a preference for collagen as a substrate. Similarly, LOXL3 has been implicated in vascular remodeling and the development of cartilage and was found to be highly expressed in mesenchymal cells, with an increased substrate affinity for collagen type XI alpha 1 and 2 (COL11A1, COL11A2) and collagen type II alpha 1 (COL2A1), the main structural component of cartilage [59]. A study by Zhang et al. (2015) showed that homozygous LOXL3 knockout mice resulted in perinatal fetal death, and LOXL3 +/– mice were found to have spinal abnormalities and cleft palate [60]. As a result, LOXL3 knockout animals could not be used for studying LOXL3 effect on cardiovascular development or changes. Lastly, LOXL2 uses collagen type IV as its preferred substrate and cross-links it through both enzymatic interactions, by the active protein, and non-enzymatic interactions, by the inactive form of the protein [61]. It is proposed to play a role in a variety of functions such as normal bone development, blood vessel stabilization, and the sprouting of new blood vessels, with LOXL2 often found localized within endothelial cells [61]. In addition, as with LOXL3, studies using LOXL2 knockout mice observed perinatal fetal death as a result of hepatic and cardiovascular defects [62]. This was confirmed by Yang et al. (2016), who demonstrated that LOXL2 secretion increased in stressed mouse hearts, triggering fibrosis [24]. Taken together, the mentioned studies implicated LOXL2 in myocardial function; however, research pertaining to LOXL2 in the heart is lacking, and thus its role in cardiac fibrosis and disease will be further discussed. Given the crucial function of LOXL2, further studies are required to understand what factors govern its regulation and function in cardiac health and disease.
2. LOXL2 in Disease
Although the LOXL2 gene has been most widely studied in cancer [50], it has recently attracted interest for its role in fibrotic diseases such as idiopathic pulmonary fibrosis and liver fibrosis during non-alcoholic steatohepatitis [63,64]. As with the previous findings by Yang and Zhao, in a study by Johnson et al. (2020), the results confirm the involvement of LOXL2 in cardiac dysfunction [65]. Nonetheless, evidence on the physiological regulation and data pertaining to the role of LOXL2 in cardiac health has not been reviewed. As such, understanding the mechanism by which LOXL2 is implicated in fibrosis during the pathogenesis of various medical conditions, like CVDs, may pave the way for future therapeutic interventions.
In a healthy state, fibroblasts are present in tissue in an inactive state. After injury, the fibroblasts are activated and differentiate into myofibroblasts in the presence of growth factors and cytokines, such as TGF-β, which stimulate ECM proteins to aid in the healing response [38]. It is known that under conditions of stress, such as inflammation, LOXL2 expression is induced, and its secretion into the extracellular space is increased [66]. Lytle et al. (2017) found, in low-density lipoprotein receptor deficient mice, that consumption of a high-fat, high-sugar diet increased plasma LOXL2 mRNA levels [67]. This was confirmed by Yang et al. (2016), who showed that augmented LOXL2 expression causes an increase in TGF-β signaling through activation of the phosphoinositide 3-kinase/protein kinase B/mechanistic target of rapamycin (PI3K/AKT/mTOR) pathway [24] (Figure 3). Furthermore, they showed that TGF-β induced myofibroblasts formation, subsequently increasing alpha smooth muscle actin (α-SMA) [24], which was previously implicated in increased collagen deposition during the fibrotic response and scar tissue formation [38,64]. In this regard, Trackman (2016) concluded that LOXL2 not only plays a role in the collagen cross-linking, but can also activate alternative fibrotic pathways through the recruitment of fibroblasts [26]. Since LOXL2 is implicated in fibrosis, it is crucial to understand its role in the pathophysiology of fibrosis-induced cardiac dysfunction.
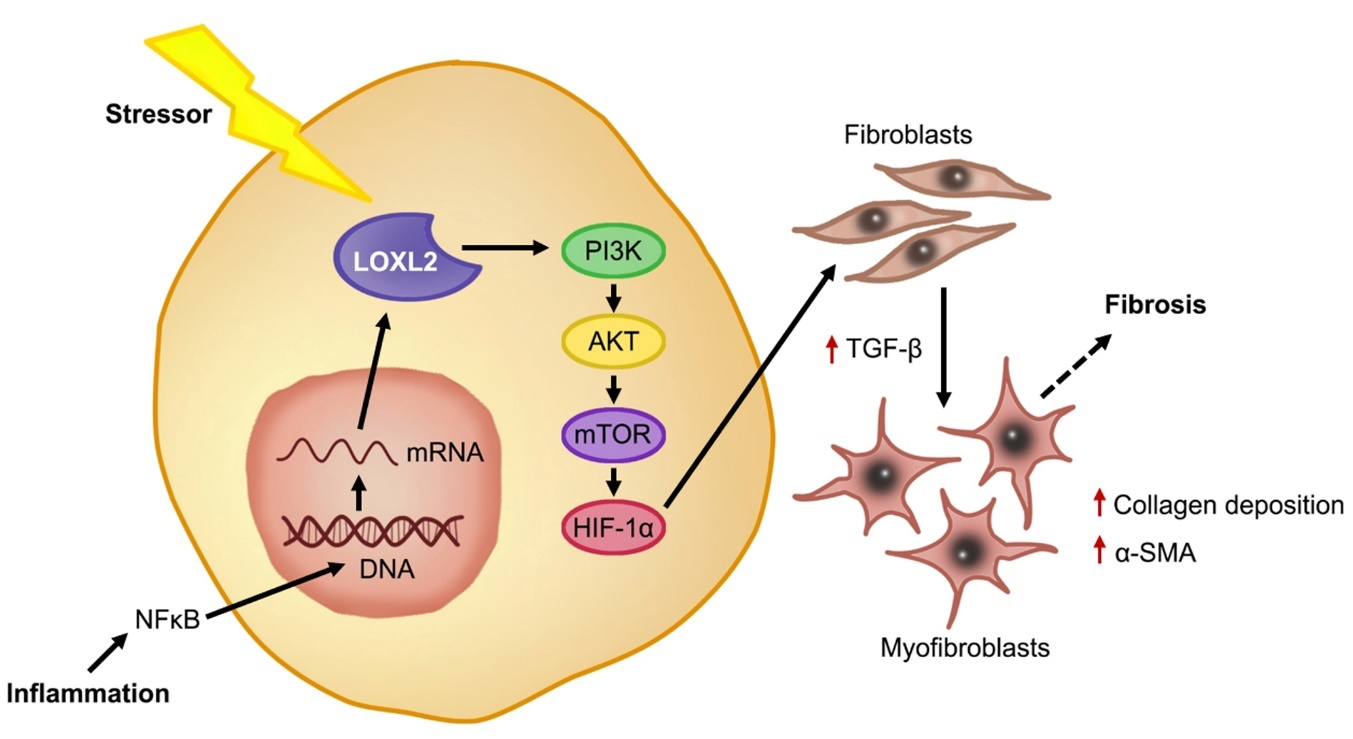
Figure 3. LOXL2-induced cardiac fibrosis. Under stress conditions such as inflammation, the activation of NFκB causes increased mRNA expression of LOXL2 and downstream, LOXL2 activates the PI3K/AKT/mTOR pathway, increasing TGF-β and triggering fibroblasts differentiation, where myofibroblasts secrete α-SMA and increase collagen deposition. Overstimulation of this process results in ECM deposition and fibrosis.
The Role of LOXL2 in the Development of Cardiovascular Disease
Diabetes is a risk factor for cardiac dysfunction and often occurs as a result of increased oxidative stress in the cells [68]. The combination of oxidative stress and the presence of high amounts of glucose in the blood leads to the production of AGEs, which can accumulate in various organs including the kidneys and heart, leading to vascular diseases and endothelial dysfunction [69–71]. AGEs are known to aggregate on proteins involved in fibrotic processes, such as fibronectin and collagen, and can alter the normal degradation of the proteins [72]. When AGEs link with collagen molecules, it has been found to decrease elasticity and increase stiffness within the vasculature and the heart tissue [73]. This leads to myocardial fibrosis by the induction of transcription factor binding, which promotes the expression of genes such as LOXL2 [73].
Cardiac fibrosis is a hallmark of cardiovascular dysfunction, where ventricular wall thickening is a consequence of excessive extracellular matrix deposition, which can reduce cardiac contractility [13]. Literature suggests that LOXL2 expression in the heart needs to be tightly regulated in order to prevent myocardial fibrosis, with these articles summarized in Table 1. Briefly, a study confirmed a positive correlation between increased LOXL2 mRNA expression and the development of cardiac dysfunction, in both transgenic mice and humans [24]. In human studies performed by Raghu et al. and Zhao et al. (2017), Raghu et al. showed that admission of Simtuzumab, which binds to LOXL2 in the treatment of fibrosis, did not improve the health outcomes of the patients with idiopathic pulmonary fibrosis [74]; however, Zhao et al. showed that patients with atrial fibrillation had increased serum LOXL2 levels, which was associated with increased left atrial size, however, there was no effect on left ventricular function [25]. In the same year, Mižíková et al. (2017) found that treating both lung fibroblasts and C57Bl/6J mice with a general inhibitor of the LOX gene family had no effect on the mRNA expression of these genes using both an in vivo and in vitro model [75]. Additionally, Craighead et al. (2018) treated hypertensive patients with the same LOX inhibitor, and found an increase in ECM-bound LOXL2 expression in these patients [76]. By means of proteomic analysis, Steppan et al. (2018) confirmed that LOXL2 mediates the stiffening of smooth muscles cells in aging using a LOXL2 knock-out mouse model [77]. This was confirmed by Torregrosa-Carrión et al. (2019), who reported that NOTCH activation led to increased expression of TGF-β2 and collagen, which form part of the LOXL2 signaling pathway [78]. Lastly, it was shown by Schilter et al. (2019) that administration of a LOXL2 inhibitor for 4-weeks, after left coronary arteries occlusion, resulted in an observed decreased myocardial fibrosis with improved cardiac output in a C57/BL6 mouse model [79]. Although there is data linking LOXL2 to fibrosis, its regulation in the fibrotic cardiac disease pathophysiology remains ill-defined, and as such, more research is required to better define LOXL2’s mechanistic role in cardiac tissue fibrosis and subsequent contractile dysfunction.
Table 1. Summary of articles investigating LOXL2 and fibrosis in cardiovascular disease.
|
Species |
Study Design |
Findings |
References |
|
Loxl2+/- knockout mice |
Mice: Underwent transaortic constriction followed by LOXL2 expression analysis and histology. |
Transgenic mice: cardiac stress results in↑ LOXL2 → myocardial fibrosis & dysfunction. Inhibition of LOXL2 activity: ↓ cardiac fibrosis and ↑ cardiac function. |
Yang et al. (2016) [24] |
|
Human |
Human: Patients presenting with HFpEF and diastolic dysfunction without symptoms underwent right-ventricular biopsies for evaluation of cardiomyopathy. |
LOXL2 acts via the PI3K/AKT pathway to activate TGF-β2. Diseased human hearts: LOXL2 ↑ in the interstitial space and serum ↑ LOXL2 expression correlated with ↑ fibrosis and myocardial dysfunction. |
|
|
Human |
Patients (aged 45-85) with idiopathic pulmonary fibrosis were treated with simtuzumab or a placebo once a week and its effects studied. |
Simtuzumab, did not improve survival rates in patients with idiopathic pulmonary fibrosis. |
Raghu et al. (2017) [74] |
|
Human |
Patients with atrial fibrillation were assessed in terms of serum LOXL2 levels, left atrial size and left ventricular function. |
Atrial fibrillation patients: ↑ serum LOXL2 Positively associated with increased left atrial size. |
Zhao et al. (2017) [25] |
|
Primary cells isolated from C57Bl/6J mice, macrophages and endothelial cells, and mouse pups |
Primary cells: cultured in the presence of a LOX inhibitor or LOX, LOXL1 and LOXL2 knocked down with siRNA. Gene expression, amine oxidase activity and microarray analyses were performed Mice: a bronchopulmonary dysplasia model was established, and lungs harvested for expression analysis. |
Lox, Loxl1, and Loxl2 are highly expressed in primary mouse lung fibroblasts. Knockdown of Lox, Loxl1, and Loxl2: associated with change in gene expression (primary mouse lung fibroblasts). BAPN: no impact on mRNA levels of LOX target-genes, in lung fibroblasts or in BAPN-treated mice. |
Mižíková et al. (2017) [75] |
|
Human |
Intradermal microdialysis fibers were placed in the forearm of young, normotensive and hypertensive individuals. Fibers treated with β-aminopropionitrile, a LOX inhibitor, or acted as a control. Norepinephrine was used to examine the vasoconstrictor function and sodium nitroprusside to study smooth muscle vasodilation. |
LOX inhibition augmented vasoconstrictor sensitivity in young and normotensive but not hypertensive patients. ECM-bound LOX expression: ↑ in hypertensive subjects vs. younger patients. Vascular stiffness & microvascular dysfunction in hypertension could be due to ↑ LOX expression. |
Craighead et al. (2018) [76] |
|
Human aortic smooth muscle cells and LOXL2+/- mice |
Human aortic smooth muscle cells were cultured and the secretome analyzed. Mice: nitric oxide production was assessed in the aortic rings. |
Proteomic analysis: LOXL2: important mediator of age-associated vascular stiffening in smooth muscle cells. Nitric oxide assessment: it ↓ LOXL2 abundance and activity in the ECM of isolated smooth muscle cells. Knock out mice: protected from age-associated vascular stiffening. Isolated aortic rings: LOXL2 mediates vascular stiffening in aging by promoting smooth muscle cell stiffness, contractility, and matrix deposition. |
Steppan et al. (2018) [77] |
|
Mouse embryonic endocardial cells, human aortic smooth muscle cells and LOXL2+/- mice |
Mouse embryonic endocardinal cells were stimulated with DLL4 and JAG1, with or without NOTCH inhibitors. Proteomics analysis of the media was conducted to identified proteins that are secreted in response to NOTCH signaling manipulation. |
Secretome analysis identified 129 factors that showed a change in expression when NOTCH was activated or repressed. NOTCH activation correlated with ↑ expression of TGF-β2 and collagen. |
Torregrosa-Carrión et al. (2019) [78] |
|
Wistar rats, Sprague Dawley rats, C57/BL6 mice |
A LOXL2/LOXL3 inhibitor, PXS-5153A, was developed and its effect on LOXL2/3 in relation to collagen cross-linking and fibrosis was assessed. |
PXS-5153A ↓ collagen cross-linking in vitro. PXS-5153A ↓ collagen expression and cross-linking, thereby ↑ liver function. In a model of myocardial infarction, addition PXS-5153A, ↑ cardiac output. This shows that inhibition of LOXL2/LOXL3 activity could be a viable treatment option for liver fibrosis. |
Schilter et al. (2019) [79] |
Symbols: → = leads to; ↓ = decreases; ↑ = increases.
3. LOXL2 Activity and Its Gene Regulatory Network
Several mechanisms of LOXL2 regulation have been proposed (Figure 4). Interestingly, galectin-3 (GAL3), although not directly related to LOXL2, is also involved in the fibrotic process through its interaction with TGF- β. Although both proinflammatory and anti-inflammatory effects have been suggested for GAL3 [80,81], it has been mostly implicated in the development and progression of HF [82,83]. LOXL2 forms part of the PI3K/AKT/mTOR pathway [24], which is able to upregulate the hypoxia-inducible factor 1 (HIF-1). This protein plays a critical role in cardiac oxygen homeostasis, and its dysregulation results in ischemic heart disease [84,85]. Nevertheless, the PI3K/AKT/mTOR pathway can activate TGF-β signaling, thereby prompting enhanced activity of the fibrotic pathways [24]. A review by Cox and Erler suggested that tissue fibrosis is caused by specific stressors, which activates TGF-β, cytokines and other inflammatory responses [86]. This in turn could result in the recruitment and subsequent activation of fibroblasts to myofibroblasts, which are then able to produce α-SMA and upregulate proteins such as connective tissue growth factor (CTGF), LOX and LOXL2 [86]. This leads to the promotion of collagen deposition and cross-linking, resulting in the modification of the ECM, tissue stiffening and organ failure [86], making LOXL2 and its gene regulatory network an ideal drug target to protect against cardiac fibrosis and improve heart function.
In addition, Chaudhury reported that signal transducers for TGF-β receptors, SMADs, are important regulators of cellular growth and development through their interaction with TGF-β [87]. Research done by Min Lu et al. (2019) on the regulation of LOX expression suggested that TGF-β1 plays a pertinent role in the phosphorylation and subsequent activation of SMAD2 and SMAD3 signaling [88], with these molecules subsequently being translocated into the nucleus where they interact with DNA-binding factors such as activating protein 1 (AP-1), specificity protein 1 (Sp-1) and NFκB [89] to have downstream effects on gene expression. LOXL2 is known to have binding sites for all three the aforementioned DNA-binding factors and thus its expression can be induced through TGF-β1/SMAD2/3 signaling [28]. Interestingly, the SMAD proteins have been found to be present in CVDs or after an ischemic cardiovascular event, where they are involved in the initiation of the fibrotic processes [89]. Moreover, when TGF is inhibited, a decrease in cardiac interstitial fibrosis as well as a suppression of late stage cardiac remodeling was observed after a myocardial infarction in mice [90]. There is also evidence in a cardiac reperfusion injury model that inhibition of the renin-angiotensin aldosterone system (RAAS) is associated with reduced levels of TGF-h and SMAD activity, together with a decrease in fibrosis [91]. Lu et al. (2019) also showed a decrease in c-Jun, one of the components of the AP-1 DNA-binding factors, caused a subsequent decrease in LOX expression as well as downstream effectors such as collagen [88]. Taken together, this suggests that TGF-β may act via SMAD and AP-1 to regulate LOX expression, increasing cardiac fibrosis. Although this proposed pathway has not yet been evaluated for LOXL2 by any single research group, it is plausible to assume, that based on the similar structure to LOX, with respect to the regulatory sites for DNA-binding factors, that LOXL2 is regulated in a similar manner to LOX. Nonetheless, in the search to better understand the role that LOXL2 plays in CVD, it is also important to understand the involvement of epigenetic modification.
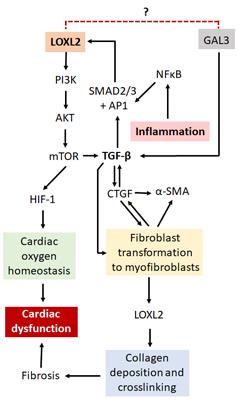
Figure 4. A proposed mechanism by which LOXL2 causes fibrosis and cardiac dysfunction. LOXL2 acts via the PI3K/AKT/mTOR pathway to activate TGF-β signaling and HIF-1 protein expression. GAL3 also activates TGF-β signaling, thus with possible similar effects as LOXL2. TGF-β signaling results in an increase in α-SMA, CTGF and LOXL2 expression, which leads to an increase in collagen deposition and cross-linking, resulting in fibrosis, ventricular stiffness and cardiac dysfunction. Inflammation causes an increase in NFκB which interacts with AP1 and Sp-1 proteins, also increasing LOXL2 expression. Dysregulation of HIF-1 protein expression results in the disruption of oxygen homeostasis in the heart, also having pathological effects. Further investigation is needed to find out whether there is a direct interaction between LOXL2 and GAL3.


