 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Murray J. Cairns | + 4876 word(s) | 4876 | 2020-08-11 11:29:17 | | | |
| 2 | Conner Chen | -1826 word(s) | 3050 | 2020-08-27 06:24:47 | | |
Video Upload Options
Schizophrenia is a severe psychiatric disorder with a complex array of signs and symptoms that causes very significant disability in young people. While schizophrenia has a strong genetic component, with heritability around 80%, there is also a very significant range of environmental exposures and stressors that have been implicated in disease development and neuropathology, such as maternal immune infection, obstetric complications, childhood trauma and cannabis exposure. It is postulated that epigenetic factors, as well as regulatory non-coding RNAs, mediate the effects of these environmental stressors. In this review, we explore the most well-known epigenetic marks, including DNA methylation and histone modification, along with emerging RNA mediators of epigenomic state, including miRNAs and lncRNAs, and discuss their collective potential for involvement in the pathophysiology of schizophrenia implicated through the postmortem analysis of brain tissue. Given that peripheral tissues, such as blood, saliva, and olfactory epithelium have the same genetic composition and are exposed to many of the same environmental exposures, we also examine some studies supporting the application of peripheral tissues for epigenomic biomarker discovery in schizophrenia. Finally, we provide some perspective on how these biomarkers may be utilised to capture a signature of past events that informs future treatment.
1. Introduction
Schizophrenia (SZ) is a debilitating psychiatric disorder that affects 0.7% of people at some point in their life. It usually occurs early in adolescence or adulthood and persists for most of an individual’s life and is recognized as one of the top 15 leading causes of disability worldwide [1]. The high heritability of disease has prompted the very substantial investigation of genetic factors through linkage studies, candidate gene studies, and more recently, genome-wide association studies (GWAS), with the hope of identifying a molecular basis for the disorder. While several family-based studies identified rare variations associated with disease development, they only contribute a small fraction of the overall population risk for this relatively common disorder. Large GWAS of schizophrenia have been more informative with the discovery of more than 140 loci [2][3]. Collectively these large GWAS highlight the genomic complexity of the syndrome with most of the risk being distributed across a large number of small effect size variants, which are highly heterogeneous between individuals. Despite the high heritability rate, estimated to be around 80%, several environmental factors have also been suggested to contribute to the development of schizophrenia. These include maternal immune activation, hypoxia, nutrient deprivation, maternal deprivation, and various toxins.
While postmortem investigations of disease-associated epigenomic changes in the brain of individuals with a diagnosis of schizophrenia have provided valuable information about the disease etiology, and supported established hypotheses about the etiology, there are several limitations for these studies, including difficult accessibility to brain samples, and therefore, small sample numbers with limited statistical power. As an alternative to studies focusing on the central nervous system, epigenetic dysregulation in peripheral tissues, such as blood, serum, saliva, and olfactory epithelium, which are more accessible in living patients, have been widely investigated. While these studies are useful for detecting biomarkers, they can also provide additional insight into disease etiology with some concordance reported between gene expression and epigenomic modification in blood and brain tissue. In this review, we have examined some of the current literature exploring changes in DNA methylation, histone modifications, and RNA mediators of epigenomic state, including microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), in the brain and peripheral tissue in schizophrenia patients and discuss their involvement in disease etiology and potential application as state and trait biomarkers of environmental exposures.
2. DNA Methylation
DNA methylation is the most stable and well-characterized epigenetic modification, which mainly occurs in cytosine and guanine-rich segments of the genome known as CpG islands. In most cases, but not exclusively, DNA methylation results in a decrease in gene expression [4]. DNA methylation changes in schizophrenia have been explored using candidate genes strategy and whole genome approaches. One of the most studied candidate genes displaying differential methylation in schizophrenia is reelin (RELN). RELN expression has been observed to be reduced between 30–50% in different areas of the brain in individuals with schizophrenia compared to healthy controls [5][6], which is highly significant given its important role in brain development and connectivity. Abdolmaleky et al. [7] were the first group who attributed RELN expression reduction to the hypermethylation of its promoter in five postmortem frontal lobe brain samples from male patients with schizophrenia compared to healthy individuals. Although similar results were reported by Grayson et al. [8], two later studies did not observe changes in the methylation status of the RELN promoter [9][10]. Glutamic acid decarboxylase 1 or GAD1 (GAD67) is another gene that has been repeatedly observed to be downregulated in postmortem analysis of the cerebral cortex in schizophrenia [11], which is shown to be associated with its promoter hypermethylation, reviewed by Guidotti et al. [12]. Similarly, COMT is a gene of interest in methylation studies of psychosis because its product, catechol-O-methyltransferase, is involved in the metabolism of dopamine, which is a major driver of positive symptoms and supports the dopamine hypothesis of schizophrenia [13]. COMT promoter methylation studies in 115 postmortem brain samples from the frontal lobe of schizophrenia patients, revealed that the promoter of the gene encoding the membrane-bound isoform is frequently hypomethylated [14]. In contrast, no schizophrenia-associated changes were observed in the frontal cortex in a cohort of 35 schizophrenia cases and 35 matched controls [10].
Some concordance for methylation status has been observed between blood and brain in relation to schizophrenia candidate genes, including RELN and COMT. The RELN promoter was observed to be hypermethylated in the peripheral blood of people with the disorder compared to the unaffected controls [15], which was consistent with postmortem methylation in the brain [7][8]. S-COMT hypermethylation was also observed in the leukocytes of schizophrenia patients (n = 177) compared to controls (n = 171) [16]. Interestingly, another study by Nohesara et al., looking at saliva as a convenient peripheral biomarker for schizophrenia, observed the hypomethylation of MB-COMT [17], which is consistent with brain studies [14].
The advance of technology has provided an opportunity for genome-wide methylation studies, called epigenome-wide association studies (EWAS). A whole-genome methylation study on the frontal cortex from 35 schizophrenia cases and 35 healthy controls reported schizophrenia-associated differences in DNA methylation in a plethora of loci, including many genes functionally related to disease etiology, such as the glutamate-receptor genes NR3B (GRIN3B) and GRIA2, and also genes involved in GABAergic neurotransmission pathways, such as MARLIN-1 (JAKMIP1) and KCNJ6 [10]. Recent studies suggest there is global DNA hypomethylation in the peripheral blood of schizophrenia patients with first-episode schizophrenia [16][18] and in discordant twins with the disorder [19]. This was supported more recently with the observation of the global hypo-methylation of peripheral leukocytes in individuals with schizophrenia [20]. A large EWAS was performed on a buffy coat of whole blood of 759 and 738 Swedish cases and controls, respectively. The top EWAS finding was located at gene FAM63B, which is part of four networks that can be associated to dopaminergic gene expression and neuronal differentiation [21]. Interestingly, a follow-up study on blood samples from bipolar disorder patients replicated FAM63B hypomethylation compared to healthy controls, confirming the possible contribution of this gene to psychiatric illness [22].
3. Histone Modifications
Histone modifications occur in four forms: histone acetylation, phosphorylation, deacetylation, methylation, with the first two, increasing and the third, decreasing gene expression. Based on the modification site, histone methylation can give rise to both expression activation and repression [4][23][24][25]. To date, little is known about histone modifications in the context of schizophrenia and this could be due to the technical difficulties associated with postmortem analyses of these marks. Tang et al. reported the correlation of gene expression levels with changes in histone H3 acetylation at promoters of four schizophrenia-related genes, including GAD67, TOMM70A, HTR2C, and PPM1E in postmortem human prefrontal cortex [26]. Huang et al. observed a decrease in the H3K4-trimethylation (H3K4me3) level in the promoter of the GAD67 gene, which was accompanied by a decrease in its expression, predominantly in female patients [27].
Reviewing the literature for histone modification studies in peripheral tissues in schizophrenia returned only one result—a pilot study, in which ChiP-seq and microarray were applied to explore genome-wide H3K4me3/H3K27me3 and gene expression, respectively, in the olfactory cells of four schizophrenia patients compared to four controls. The authors detected 22 genes for which schizophrenia-associated changes in expression were likely to be linked with changes in histone trimethylation. qPCR validated expression changes for three genes, MGST1, DAAM2, and LPXN [28].
4. MicroRNA (miRNA)
MicroRNA (miRNA) are a class of small non-coding RNA which are approximately 22 nucleotides of length. Through their 5′-end seed region, miRNA can bind to a target sequence on the 3′-UTR of mature mRNAs and lead to either the degradation of mRNA or repression of its translation (Figure 1). As each miRNA needs only partial homology/complementarity to their target mRNAs, to achieve post-transcriptional silencing, they can each affect the expression of hundreds of genes simultaneously and synchronize multiple components of independent signaling pathways [29].
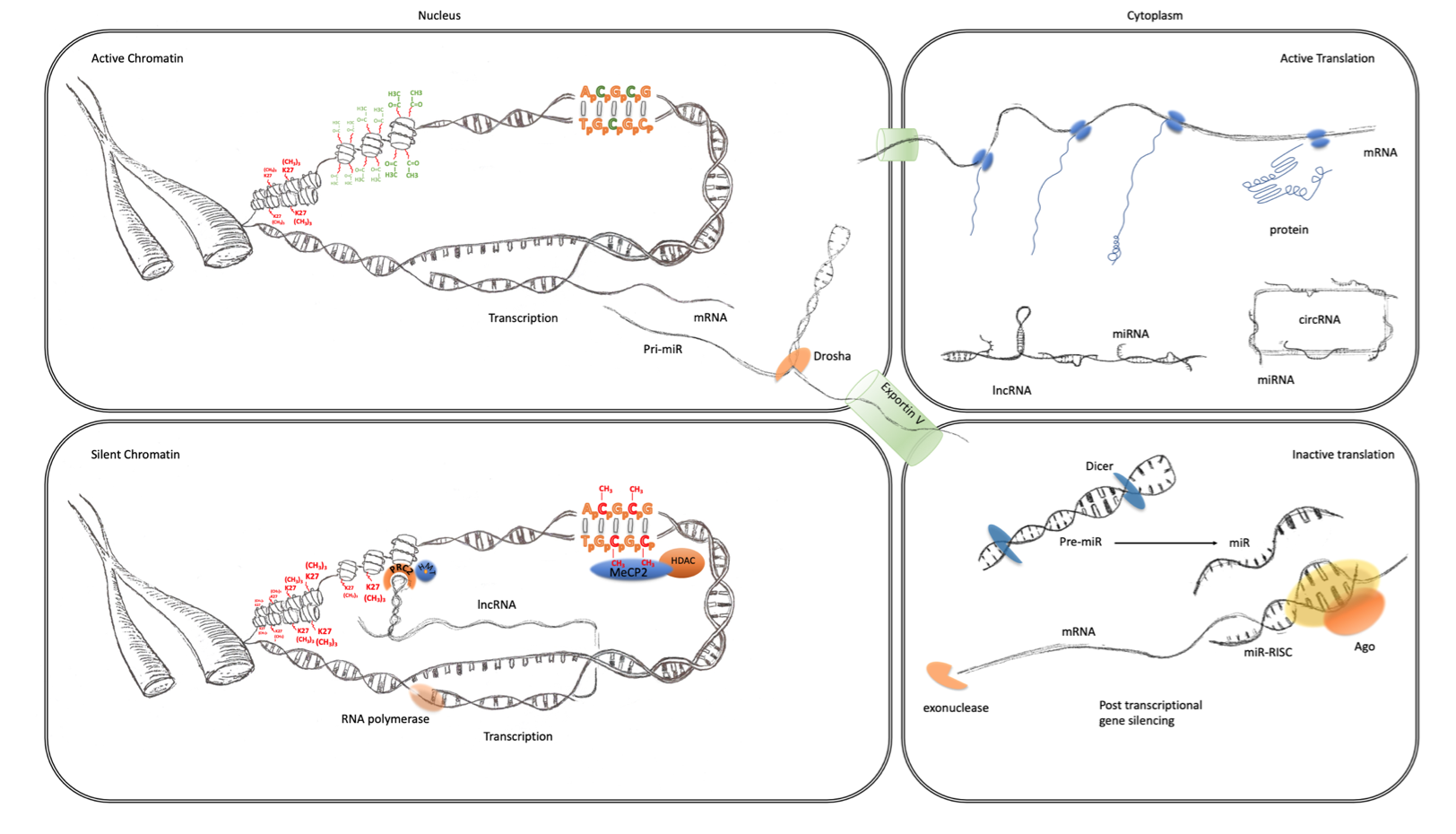
Figure 1. Transcriptional and post-transcriptional regulation of gene expression. The four panels of this schematic are representative of the major epigenomic mechanisms’ associated active and inactive states in the nucleus and cytoplasm. Active chromatin (top left) characterized by an expanded or open euchromatin domain is established by the depletion of histone H3 lysing 27 trimethylation (K27(CH3)3 red) and an increase in histone H3 and H4 acetylation (green). This gives rise to mRNA transcription and non-coding RNA, such as primary miRNA transcripts. After interaction with the microprocessor complex (Drosha/DGCR8), the processed pre-miR are exported to the cytoplasm via exportin 5. mRNA is also exported to the cytoplasm (top right panel) where it becomes associated with ribosomes and translated into protein (blue). This is facilitated by the suppression of potentially active miRNA, which are sequestered through interaction with lincRNA and circRNA. Inactive chromatin (bottom left) is associated with cytosine methylation (red), which is particularly important at CpG islands and promoters where they can bind to MeCP2 which can recruit histone de-acetylases (HDAC). These reduce histone acetylation, which leads to greater compaction of chromatin into a state known as heterochromatin. LincRNA transcripts can also form a scaffold for the assembly of a ribonucleoprotein complex, including the PRC2, that directs the activity of histone methyltransferases (HMTs) that catalyze the trimethylation of histone H3 lysine 27. This modification also further enhances the contraction of chromatin into inactive heterochromatin. Post-transcriptional regulation of gene expression (bottom right panel) can also be facilitated by the maturation of pre-miRs through the activity of dicer. The mature miRNAs guide the RNA-induced silencing complex (comprising Argonaute proteins and other co-factors) to the 3′UTR of their cognate mRNA, causing translational repression and degradation through exonuclease activity.
miRNAs are believed to have a crucial role in the development of central nervous system, and several investigations have now observed changes in their expression profile in the postmortem brains of subjects with schizophrenia. Our group initially investigated miRNA expression in postmortem superior temporal gyrus (STG) from 21 subjects with the disorder and 21 non-psychiatric controls and observed the up regulation of the brain-enriched miRNA, miR-181b [30]. After further analysis and the addition of the dorsolateral prefrontal cortex (BA9), we revealed that a larger proportion of miRNA were elevated, suggesting a global increase in miRNA biogenesis [31]. More recently we integrated differentially expressed miRNA and mRNA from postmortem DLPFC (BA46) in a cohort of 37 matched pairs of case/control SZ subjects, and observed substantial correlation networks between miR-92a, miR-495, and miR-134, and their target genes in pathways involved in neurodevelopment and oligodendrocyte function [32].
With such a substantial change in miRNA expression observed in the central nervous system in schizophrenia, it is plausible that similar changes take place in peripheral tissue and may be accessible biomarkers of related disease processes. This hypothesis was supported by the work of Lai et al. in their report of a seven-miRNA signature in peripheral blood mononuclear cells (PBMCs) of SZ patients [33]. Interestingly, the most differentially expressed miRNA was miR-34a, whose applicability as a biomarker with high diagnostic sensitivity and specificity was later confirmed in a meta-analysis on PBMCs [34]. In our laboratory, PBMCs from 112 schizophrenia patients and 78 controls were examined and we identified the downregulation of 34 microRNAs at FDR = 0, three of which, including miR-134, miR-128 and miR-181b, are brain enriched. Interestingly, of the most significantly downregulated miRNAs, 17, including miR-134, are clustered in two closely neighboring positions on chromosome 14. Notably, this locus, known as the DLK1-DIO3 region, is imprinted such that the associated miRNA cluster is only expressed from the maternal chromosome. This, alongside the observation that no CNV had occurred in this region in a subset of samples including 57 controls and 81 cases, implicated the plausible epigenetic regulation of this cluster expression [35]. Serum miRNAs, referred to as circulating miRNAs, have also been observed to be altered in the disorder. In the first study on serum miRNAs expression in schizophrenia [36], with 115 patients and 40 controls, seven molecules were identified as potential biomarkers, including miR181b, whose dysregulation in superior temporal gyrus [30] and PBMCs [35] had been previously reported by our group. More recently, circulating miRNAs were profiled in a test cohort of 164 schizophrenia patients and 187 control subjects. The captured miRNAs were then validated by qPCR in an independent cohort of 400 schizophrenia patients, 213 control subjects, and 162 patients with non-schizophrenia psychiatric disorders. Plasma miRNA screening identified eight upregulated miRNAs in schizophrenia, with qPCR analysis supporting the upregulation of miR-130b and miR-193a-3p in schizophrenia but not in other disorders tested, suggesting that these two miRNAs could be used to develop a diagnostic tool for schizophrenia [37].
The up-regulation of miR-137 in peripheral tissues in schizophrenia was reported in three independent studies, suggesting it may have applications as a biomarker [34][38][39]. miR-137 is a brain-enriched miRNA predicted to target over 1000 genes involved in various pathways, such as cell cycle, neural development, proliferation, and differentiation. Experimental evidence supports the involvement of miR-137 in the regulation of adult neurogenesis, dendritic development, and neuronal maturation [40]. Its involvement in schizophrenia was supported by the genome-wide association of almost 37,000 cases and 113,000 controls, with the second most associated variant in proximity to the MIR137 gene (rs1702294, p = 3.4 × 10−19) [2].
5. Long Non-Coding RNAs (lncRNAs)
Long non-coding RNAs (lncRNAs) are non-protein coding transcripts of greater than 200 nucleotides in length and functionally can regulate gene expression at both transcription and post-transcription levels [41] (Figure 1). Barry et al. showed that the brain-enriched long non-coding RNA Gomafu interacts with several splicing factor proteins, such as SF1 and QKI, and takes part in the alternative splicing of DISC1 and ERBB4, both involved in schizophrenia etiology. It was significantly downregulated in response to depolarization in mouse primary neurons and human-induced pluripotent stem cells (hiPSCs). In addition, they reported a 1.75-fold reduction in Gomafu expression in the STG of 28 schizophrenia subjects relative to 28 controls [42]. Hu et al. identified the differential expression of 35 lncRNAs in two brain regions, some of which were related to neuron ensheathment, metabolic processes, myelination, and oligodendrocyte differentiation [43]. More than 200 lncRNAs were also reported to be differentially expressed in postmortem amygdala from 22 individuals with schizophrenia compared to 24 control subjects, three of which, AC005009.2, RP11-724N1.1, and RP11-677M14.2, were located in schizophrenia-associated loci [44].
Ren et al. examined PBMCs from a cohort consisting of 19 first-episode early-onset schizophrenia (EOS) patients and 18 controls. Instead of differential expression analysis at the level of individual genes, they applied weighted gene co-expression network analysis (WGCNA) to identify higher order relationships among gene products. Their analysis revealed two schizophrenia-associated lncRNA modules. They did the same analysis for mRNA which returned three disease-associated mRNA modules and then combined the two datasets through canonical correlation analysis (CCA) and observed the disturbance of mitochondria, which is consistent with other studies, indicating the role of mitochondrial dysfunction in schizophrenia development [45].
While the search for clinically useful diagnostic biomarkers for schizophrenia has a history spanning several decades, there is little in the way of successful outcomes to report for several reasons. Perhaps the most important of these is the existence of significant levels of heterogeneity which, in some cases, can be reduced to different subtypes for many of these disorders, which is reflected in the high levels of variation in gene expression or chromatin modification between tissues and patient populations [46]. While there has been great progress towards covering the common variant genetic heterogeneity through large mega GWAS and meta-analysis, there have been no comprehensive meta-analyses on epigenetic diagnostic biomarker studies. Some of the non-epigenetic diagnostic biomarkers suggested for SZ, which have received support from meta-analyses, include decreased levels of pyridoxal (Vitamin B6) and nerve growth factor (NGF), and soluble interleukin-(IL)-2 receptor (sIL-2R) level increase, confirmed by a recent umbrella review [47], as well as a significantly increased level of the acute-phase protein C-Reactive Protein (CRP) [48][. On the other hand, pinpointing prognostic biomarkers, that can predict the future risk of developing schizophrenia before the onset of disease symptoms, is of critical importance. This is not convenient and time-efficient in human studies and, therefore, developmental animal models are required, through which the effects of perinatal and/or early postnatal exposure to environmental manipulations and/or drug administration are investigated in peripheral tissues over the course of development [49]. Our literature search for these types of studies returned only one result, in which pregnant mice were treated with the toxicant bisphenol A (BPA), that disturbs neurodevelopment with long-term effects on behavior, and the offspring went through gene expression and DNA methylation analyses in the hippocampus and blood. Based on the results, the authors suggested that BDNF DNA methylation in the blood can predict early life adversity-induced epigenetic changes in the brain and may be a novel clinical epigenetic biomarker to predict the risk for psychopathology [50].
To conclude, epigenomic features and associated non-coding RNA provide an important mechanism for the neuropathology of schizophrenia that, in some cases, can be tracked through peripheral tissues. Where these provide important information/biomarkers that inform on an individual’s trait and state risk, they may have utility to both facilitate early intervention and direct intervention in support of precision medicine strategies. Future work that better captures and integrates these forms of information through poly-omic methodologies will make an important contribution toward this aim by providing the training sets needed to establish an unbiased etiological framework. While much of this exploration is focused specifically on disease, more studies using alternative phenotypic labels related to sub-types or other endophenotypes will further refine our models and provide the basis for more cross-disorder integration of psychiatric and behavioral syndromes.
References
- Collaborators, G.D.a.I.I.a.P. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211-1259, doi:10.1016/s0140-6736(17)32154-2.
- Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421-427, doi:10.1038/nature13595.
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L., et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 2018, 50, 381-389, doi:10.1038/s41588-018-0059-2.
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457-463, doi:10.1038/nature02625.
- Impagnatiello, F.; Guidotti, A.R.; Pesold, C.; Dwivedi, Y.; Caruncho, H.; Pisu, M.G.; Uzunov, D.P.; Smalheiser, N.R.; Davis, J.M.; Pandey, G.N., et al. A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc Natl Acad Sci U S A 1998, 95, 15718-15723, doi:10.1073/pnas.95.26.15718.
- Guidotti, A.; Auta, J.; Davis, J.M.; Di-Giorgi-Gerevini, V.; Dwivedi, Y.; Grayson, D.R.; Impagnatiello, F.; Pandey, G.; Pesold, C.; Sharma, R., et al. Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: a postmortem brain study. Arch Gen Psychiatry 2000, 57, 1061-1069, doi:10.1001/archpsyc.57.11.1061.
- Abdolmaleky, H.M.; Cheng, K.H.; Russo, A.; Smith, C.L.; Faraone, S.V.; Wilcox, M.; Shafa, R.; Glatt, S.J.; Nguyen, G.; Ponte, J.F., et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005, 134b, 60-66, doi:10.1002/ajmg.b.30140.
- Grayson, D.R.; Jia, X.; Chen, Y.; Sharma, R.P.; Mitchell, C.P.; Guidotti, A.; Costa, E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A 2005, 102, 9341-9346, doi:10.1073/pnas.0503736102.
- Tochigi, M.; Iwamoto, K.; Bundo, M.; Komori, A.; Sasaki, T.; Kato, N.; Kato, T. Methylation status of the reelin promoter region in the brain of schizophrenic patients. Biol Psychiatry 2008, 63, 530-533, doi:10.1016/j.biopsych.2007.07.003.
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A., et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am J Hum Genet 2008, 82, 696-711, doi:10.1016/j.ajhg.2008.01.008.
- Mitchell, A.C.; Jiang, Y.; Peter, C.; Akbarian, S. Transcriptional regulation of GAD1 GABA synthesis gene in the prefrontal cortex of subjects with schizophrenia. Schizophr Res 2015, 167, 28-34, doi:10.1016/j.schres.2014.10.020.
- Guidotti, A.; Auta, J.; Chen, Y.; Davis, J.M.; Dong, E.; Gavin, D.P.; Grayson, D.R.; Matrisciano, F.; Pinna, G.; Satta, R., et al. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 2011, 60, 1007-1016, doi:10.1016/j.neuropharm.2010.10.021.
- Nishioka, M.; Bundo, M.; Kasai, K.; Iwamoto, K. DNA methylation in schizophrenia: progress and challenges of epigenetic studies. Genome Med 2012, 4, 96, doi:10.1186/gm397.
- Abdolmaleky, H.M.; Cheng, K.H.; Faraone, S.V.; Wilcox, M.; Glatt, S.J.; Gao, F.; Smith, C.L.; Shafa, R.; Aeali, B.; Carnevale, J., et al. Hypomethylation of MB-COMT promoter is a major risk factor for schizophrenia and bipolar disorder. Hum Mol Genet 2006, 15, 3132-3145, doi:10.1093/hmg/ddl253.
- Nabil Fikri, R.M.; Norlelawati, A.T.; Nour El-Huda, A.R.; Hanisah, M.N.; Kartini, A.; Norsidah, K.; Nor Zamzila, A. Reelin (RELN) DNA methylation in the peripheral blood of schizophrenia. J Psychiatr Res 2017, 88, 28-37, doi:10.1016/j.jpsychires.2016.12.020.
- Melas, P.A.; Rogdaki, M.; Ösby, U.; Schalling, M.; Lavebratt, C.; Ekström, T.J. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. Faseb j 2012, 26, 2712-2718, doi:10.1096/fj.11-202069.
- Nohesara, S.; Ghadirivasfi, M.; Mostafavi, S.; Eskandari, M.R.; Ahmadkhaniha, H.; Thiagalingam, S.; Abdolmaleky, H.M. DNA hypomethylation of MB-COMT promoter in the DNA derived from saliva in schizophrenia and bipolar disorder. J Psychiatr Res 2011, 45, 1432-1438, doi:10.1016/j.jpsychires.2011.06.013.
- Nishioka, M.; Bundo, M.; Koike, S.; Takizawa, R.; Kakiuchi, C.; Araki, T.; Kasai, K.; Iwamoto, K. Comprehensive DNA methylation analysis of peripheral blood cells derived from patients with first-episode schizophrenia. J Hum Genet 2013, 58, 91-97, doi:10.1038/jhg.2012.140.
- Bönsch, D.; Wunschel, M.; Lenz, B.; Janssen, G.; Weisbrod, M.; Sauer, H. Methylation matters? Decreased methylation status of genomic DNA in the blood of schizophrenic twins. Psychiatry Res 2012, 198, 533-537, doi:10.1016/j.psychres.2011.09.004.
- Murata, Y.; Ikegame, T.; Koike, S.; Saito, T.; Ikeda, M.; Sasaki, T.; Iwata, N.; Kasai, K.; Bundo, M.; Iwamoto, K. Global DNA hypomethylation and its correlation to the betaine level in peripheral blood of patients with schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2020, 99, 109855, doi:10.1016/j.pnpbp.2019.109855.
- Aberg, K.A.; McClay, J.L.; Nerella, S.; Clark, S.; Kumar, G.; Chen, W.; Khachane, A.N.; Xie, L.; Hudson, A.; Gao, G., et al. Methylome-wide association study of schizophrenia: identifying blood biomarker signatures of environmental insults. JAMA Psychiatry 2014, 71, 255-264, doi:10.1001/jamapsychiatry.2013.3730.
- Starnawska, A.; Demontis, D.; McQuillin, A.; O'Brien, N.L.; Staunstrup, N.H.; Mors, O.; Nielsen, A.L.; Børglum, A.D.; Nyegaard, M. Hypomethylation of FAM63B in bipolar disorder patients. Clin Epigenetics 2016, 8, 52, doi:10.1186/s13148-016-0221-6.
- Gavin, D.P.; Sharma, R.P. Histone modifications, DNA methylation, and schizophrenia. Neurosci Biobehav Rev 2010, 34, 882-888, doi:10.1016/j.neubiorev.2009.10.010.
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell 2010, 38, 662-674, doi:10.1016/j.molcel.2010.03.021.
- Lai, F.; Orom, U.A.; Cesaroni, M.; Beringer, M.; Taatjes, D.J.; Blobel, G.A.; Shiekhattar, R. Activating RNAs associate with Mediator to enhance chromatin architecture and transcription. Nature 2013, 494, 497-501, doi:10.1038/nature11884.
- Tang, B.; Dean, B.; Thomas, E.A. Disease- and age-related changes in histone acetylation at gene promoters in psychiatric disorders. Transl Psychiatry 2011, 1, e64, doi:10.1038/tp.2011.61.
- Huang, H.S.; Matevossian, A.; Whittle, C.; Kim, S.Y.; Schumacher, A.; Baker, S.P.; Akbarian, S. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. J Neurosci 2007, 27, 11254-11262, doi:10.1523/jneurosci.3272-07.2007.
- Kano, S.; Colantuoni, C.; Han, F.; Zhou, Z.; Yuan, Q.; Wilson, A.; Takayanagi, Y.; Lee, Y.; Rapoport, J.; Eaton, W., et al. Genome-wide profiling of multiple histone methylations in olfactory cells: further implications for cellular susceptibility to oxidative stress in schizophrenia. Mol Psychiatry 2013, 18, 740-742, doi:10.1038/mp.2012.120.
- Geaghan, M.; Cairns, M.J. MicroRNA and Posttranscriptional Dysregulation in Psychiatry. Biol Psychiatry 2015, 78, 231-239, doi:10.1016/j.biopsych.2014.12.009.
- Beveridge, N.J.; Tooney, P.A.; Carroll, A.P.; Gardiner, E.; Bowden, N.; Scott, R.J.; Tran, N.; Dedova, I.; Cairns, M.J. Dysregulation of miRNA 181b in the temporal cortex in schizophrenia. Hum Mol Genet 2008, 17, 1156-1168, doi:10.1093/hmg/ddn005.
- Beveridge, N.J.; Gardiner, E.; Carroll, A.P.; Tooney, P.A.; Cairns, M.J. Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol Psychiatry 2010, 15, 1176-1189, doi:10.1038/mp.2009.84.
- Santarelli, D.M.; Carroll, A.P.; Cairns, H.M.; Tooney, P.A.; Cairns, M.J. Schizophrenia-associated MicroRNA-Gene Interactions in the Dorsolateral Prefrontal Cortex. Genomics Proteomics Bioinformatics 2019, 17, 623-634, doi:10.1016/j.gpb.2019.10.003.
- Lai, C.Y.; Yu, S.L.; Hsieh, M.H.; Chen, C.H.; Chen, H.Y.; Wen, C.C.; Huang, Y.H.; Hsiao, P.C.; Hsiao, C.K.; Liu, C.M., et al. MicroRNA expression aberration as potential peripheral blood biomarkers for schizophrenia. PLoS One 2011, 6, e21635, doi:10.1371/journal.pone.0021635.
- Liu, S.; Zhang, F.; Wang, X.; Shugart, Y.Y.; Zhao, Y.; Li, X.; Liu, Z.; Sun, N.; Yang, C.; Zhang, K., et al. Diagnostic value of blood-derived microRNAs for schizophrenia: results of a meta-analysis and validation. Sci Rep 2017, 7, 15328, doi:10.1038/s41598-017-15751-5.
- Gardiner, E.; Beveridge, N.J.; Wu, J.Q.; Carr, V.; Scott, R.J.; Tooney, P.A.; Cairns, M.J. Imprinted DLK1-DIO3 region of 14q32 defines a schizophrenia-associated miRNA signature in peripheral blood mononuclear cells. Mol Psychiatry 2012, 17, 827-840, doi:10.1038/mp.2011.78.
- Shi, W.; Du, J.; Qi, Y.; Liang, G.; Wang, T.; Li, S.; Xie, S.; Zeshan, B.; Xiao, Z. Aberrant expression of serum miRNAs in schizophrenia. J Psychiatr Res 2012, 46, 198-204, doi:10.1016/j.jpsychires.2011.09.010.
- Wei, H.; Yuan, Y.; Liu, S.; Wang, C.; Yang, F.; Lu, Z.; Wang, C.; Deng, H.; Zhao, J.; Shen, Y., et al. Detection of circulating miRNA levels in schizophrenia. Am J Psychiatry 2015, 172, 1141-1147, doi:10.1176/appi.ajp.2015.14030273.
- Ma, J.; Shang, S.; Wang, J.; Zhang, T.; Nie, F.; Song, X.; Heping, Z.; Zhu, C.; Zhang, R.; Hao, D. Identification of miR-22-3p, miR-92a-3p, and miR-137 in peripheral blood as biomarker for schizophrenia. Psychiatry Res 2018, 265, 70-76, doi:10.1016/j.psychres.2018.03.080.
- Wu, S.; Zhang, R.; Nie, F.; Wang, X.; Jiang, C.; Liu, M.; Valenzuela, R.K.; Liu, W.; Shi, Y.; Ma, J. MicroRNA-137 Inhibits EFNB2 Expression Affected by a Genetic Variant and Is Expressed Aberrantly in Peripheral Blood of Schizophrenia Patients. EBioMedicine 2016, 12, 133-142, doi:10.1016/j.ebiom.2016.09.012.
- Mahmoudi, E.; Cairns, M.J. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry 2017, 22, 44-55, doi:10.1038/mp.2016.150.
- Barry, G. Integrating the roles of long and small non-coding RNA in brain function and disease. Mol Psychiatry 2014, 19, 410-416, doi:10.1038/mp.2013.196.
- Barry, G.; Briggs, J.A.; Vanichkina, D.P.; Poth, E.M.; Beveridge, N.J.; Ratnu, V.S.; Nayler, S.P.; Nones, K.; Hu, J.; Bredy, T.W., et al. The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry 2014, 19, 486-494, doi:10.1038/mp.2013.45.
- Hu, J.; Xu, J.; Pang, L.; Zhao, H.; Li, F.; Deng, Y.; Liu, L.; Lan, Y.; Zhang, X.; Zhao, T., et al. Systematically characterizing dysfunctional long intergenic non-coding RNAs in multiple brain regions of major psychosis. Oncotarget 2016, 7, 71087-71098, doi:10.18632/oncotarget.12122.
- Liu, Y.; Chang, X.; Hahn, C.G.; Gur, R.E.; Sleiman, P.A.M.; Hakonarson, H. Non-coding RNA dysregulation in the amygdala region of schizophrenia patients contributes to the pathogenesis of the disease. Transl Psychiatry 2018, 8, 44, doi:10.1038/s41398-017-0030-5.
- Ren, Y.; Cui, Y.; Li, X.; Wang, B.; Na, L.; Shi, J.; Wang, L.; Qiu, L.; Zhang, K.; Liu, G., et al. A co-expression network analysis reveals lncRNA abnormalities in peripheral blood in early-onset schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry 2015, 63, 1-5, doi:10.1016/j.pnpbp.2015.05.002.
- Peedicayil, J. Identification of Biomarkers in Neuropsychiatric Disorders Based on Systems Biology and Epigenetics. Front Genet 2019, 10, 985, doi:10.3389/fgene.2019.00985.
- Carvalho, A.F.; Solmi, M.; Sanches, M.; Machado, M.O.; Stubbs, B.; Ajnakina, O.; Sherman, C.; Sun, Y.R.; Liu, C.S.; Brunoni, A.R., et al. Evidence-based umbrella review of 162 peripheral biomarkers for major mental disorders. Transl Psychiatry 2020, 10, 152, doi:10.1038/s41398-020-0835-5.
- Fernandes, B.S.; Steiner, J.; Bernstein, H.G.; Dodd, S.; Pasco, J.A.; Dean, O.M.; Nardin, P.; Gonçalves, C.A.; Berk, M. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry 2016, 21, 554-564, doi:10.1038/mp.2015.87.
- Winship, I.R.; Dursun, S.M.; Baker, G.B.; Balista, P.A.; Kandratavicius, L.; Maia-de-Oliveira, J.P.; Hallak, J.; Howland, J.G. An Overview of Animal Models Related to Schizophrenia. Can J Psychiatry 2019, 64, 5-17, doi:10.1177/0706743718773728.
- Kundakovic, M.; Gudsnuk, K.; Herbstman, J.B.; Tang, D.; Perera, F.P.; Champagne, F.A. DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci U S A 2015, 112, 6807-6813, doi:10.1073/pnas.1408355111.

