 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ewelina Klupczyńska | + 4137 word(s) | 4137 | 2021-12-20 03:49:31 | | | |
| 2 | Ewelina Klupczyńska | Meta information modification | 4137 | 2021-12-20 11:56:45 | | | | |
| 3 | Peter Tang | Meta information modification | 4137 | 2021-12-21 03:35:51 | | |
Video Upload Options
Methylation occurs in both plants and animals and is an important epigenetic modification due to its role in gene regulation, transposable element (TE) silencing, chromosomal interactions, and stability of the plant genome. Epigenetic modifications, including chromatin modifications and DNA methylation, play key roles in regulating gene expression in both plants and animals. Transmission of epigenetic markers is important for some genes to maintain specific expression patterns and preserve the status quo of the cell. The implications of epigenetics are important for adaptation and phenotypic plasticity because they provide the potential for tree conservation in forest ecosystems exposed to adverse conditions resulting from global warming and regional climate fluctuations.
1. Introduction
2. Effects of DNA Methylation of Forest Trees on Gene Expression and Climate Adaptation
2.1. DNA methylation in Plants

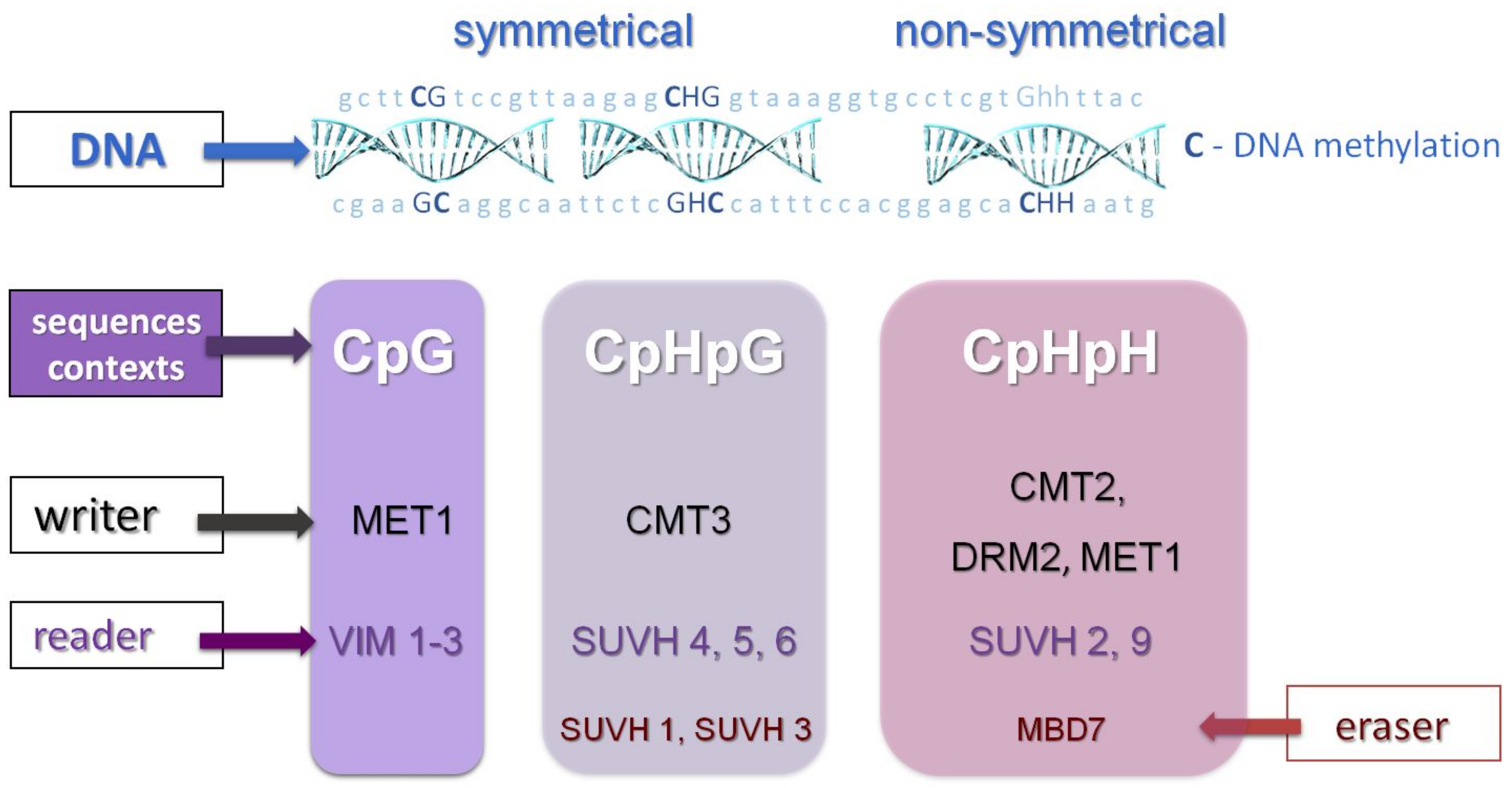
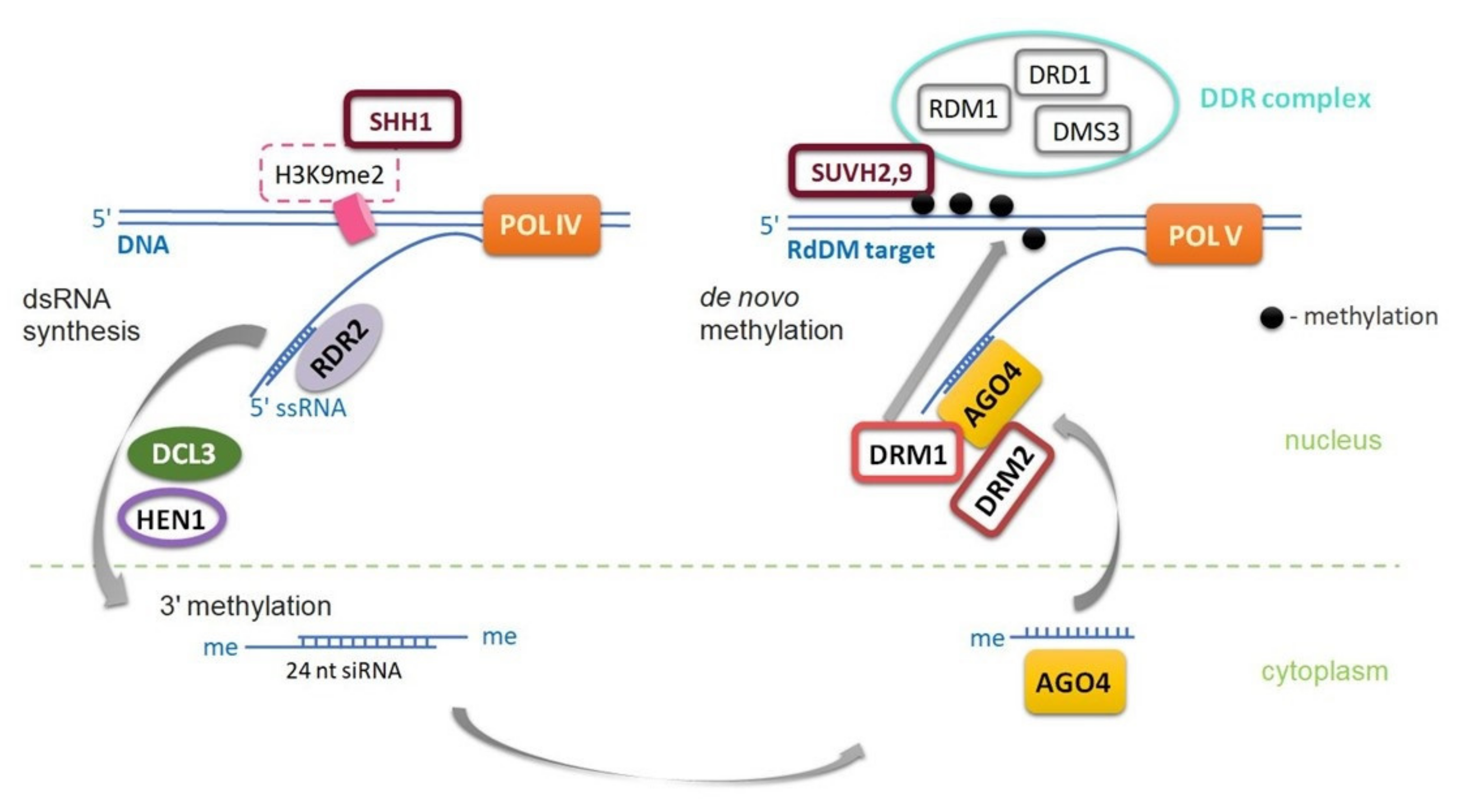
Maintenance of DNA methylation in the CpHpG context requires chromomethylase 3 (CMT3) activity. Methylation in the CpHpH context requires chromomethylase 2 (CMT2) activity [17][18], and a large amount of CpHpH methylation is maintained by domains rearranged methyltransferase 2 (DRM2) in the RNA-dependent DNA methylation (RdDM) pathway [15][19][20][21], which is also responsible for de novo methylation in all three sequence contexts [8][9][18]. CpHpH methylation is asymmetric, meaning methylation will be lost in one progeny strand. Chromomethylases contain both a chromodomain and a DNA methyltransferase domain and interact with some proteins (suppressor of variegation su(var) homolog, SUVH) to ensure proper deposition of histone H3K9 (H3K9me2) methylation, as well as CpHpG or CpHpH in transposable elements (TEs) [20]. SUVH proteins are essential for accessing the regulatory mechanisms of genes located in close proliferating transposable elements (TEs) [20]. DRM2 and MET1 proteins share significant homology with mammalian methyltransferases (DNMT3 and DNMT1). The CMT3 protein is unique to plants and belongs to a family of chromomethylases that are both “readers” of histone methylation and “writers” of DNA methylation [20][22]. Most methylation in plants occurs in transposable elements (TEs), but also in the bodies of active genes where it is restricted to the CpG context [9][18]. The first step in de novo DNA methylation pattern formation is the RNA-dependent DNA methylation pathway, which relies on specialized, plant-specific RNA polymerases POL IV or POL V (POL VI is also specific for grasses) [20][22] (Figure 3).
2.2. Forest Trees—Ecosystems Important to Humans
Forests cover approximately 31% of the world’s land area, or 4 billion hectares (http://www.fao.org/state-of-forests/en, accessed on 19 July 2021). Almost half of these are intact (natural forests), and more than one-third are naturally regenerating forests of native species where there are no traces of human activities and natural ecological processes are undisturbed (primary forests). Forests are dynamic ecosystems with high environmental, social, and economic importance [1]. However, the global changes currently occurring on Earth, such as desertification, insect invasion, abiotic stresses, deforestation, degradation, and climate change, pose a significant threat to the condition of forests.
2.3. Effects of DNA Methylation on Adaptations of Forest Trees
2.4. Epigenetic Modifications of Trees and Environmental Conditions—A Review of Existing Research and the Current State of Knowledge
Analysis of the white poplar Populus alba L. DNA methylation profiles from vegetatively propagated populations [86] showed that environmental conditions strongly influence internal cytosine hemimethylation. Eighty-three samples of white poplar at different locations in Sardinia were investigated by MSAP. The analysis was performed on genomic DNA extracted from leaves at the same juvenile stage. The study showed that the genetic biodiversity of poplar is quite limited but is balanced by epigenetic interpopulation molecular variation. The results clearly showed that ramets of the same clone were differentially methylated according to geographical location. In poplar, epigenetic changes are frequent and occur more rapidly in response to environmental stimuli, confirming the molecular model of stress epigenetic memory for plant responses to stress leading to increased overall methylation levels induced by external stimuli [87].
The relationship between environmental adaptation and DNA methylation has also been shown in studies on natural populations of the holm oak Quercus ilex L. of Mediterranean forests [88]. Methylation patterns and levels were assessed in individuals from control forest plots (in southern Catalonia, Spain) and in individuals experiencing drought stress (exposed to several years of drought at levels projected for decades to come). Drought-exposed plants had a percentage of hypermethylated loci lower than the control, while the percentage of fully methylated loci was significantly higher. These results also demonstrate that changes in DNA methylation contribute greatly to the ability of trees to rapidly acclimate to changing environmental conditions.
Nevertheless, this is only the beginning of a full understanding of the function and operation of the epigenome. Many of the processes that occur during epigenetic modifications and the modifications themselves that have been most thoroughly understood in model plants (also mentioned above) have not been confirmed in studies of forest trees. Therefore, there is still a long way to go to fully understand the function of epigenetic modifications in trees in the context of both abiotic stresses and strictly global climate change.
References
- Amaral, J.; Ribeyre, Z.; Vigneaud, J.; Sow, M.D.; Fichot, R.; Messier, C.; Pinto, G.; Nolet, P.; Maury, S. Advances and Promises of Epigenetics for Forest Trees. Forests 2020, 11, 976.
- Johnson, T.B.; Coghill, R.D. Researches on Pyrimidines. C111. The Discovery of 5-Methyl-Cytosine in Tuberculinic Acid, The Nucleic Acid of the Tubercle bacillus. J. Am. Chem. Soc. 1925, 47, 2838–2844.
- Waddington, C.H. An Introduction to Modern Genetics; The Macmillan Company: New York, NY, USA, 1939.
- Yakovlev, I.; Fossdal, C.G.; Skrøppa, T.; Olsen, J.E.; Jahren, A.H.; Johnsen, Ø. An Adaptive Epigenetic Memory in Conifers with Important Implications for Seed Production. Seed Sci. Res. 2012, 22, 63–76.
- Alakärppä, E.; Salo, H.M.; Valledor, L.; Cañal, M.J.; Häggman, H.; Vuosku, J. Natural Variation of DNA Methylation and Gene Expression May Determine Local Adaptations of Scots Pine Populations. J. Exp. Bot. 2018, 69, 5293–5305.
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis M6A Demethylase Activity Modulates Viral Infection of a Plant Virus and the M6A Abundance in Its Genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760.
- Jones, P.A.; Takai, D. The Role of DNA Methylation in Mammalian Epigenetics. Science 2001, 293, 1068–1070.
- Matzke, M.; Kanno, T.; Daxinger, L.; Huettel, B.; Matzke, A.J. RNA-Mediated Chromatin-Based Silencing in Plants. Curr. Opin. Cell Biol. 2009, 21, 367–376.
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220.
- Illingworth, R.S.; Bird, A.P. CpG Islands—‘A Rough Guide’. FEBS Lett. 2009, 583, 1713–1720.
- Li, S.; Tollefsbol, T.O. DNA Methylation Methods: Global DNA Methylation and Methylomic Analyses. Methods 2021, 187, 28–43.
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17.
- Meyer, P. Epigenetic Variation and Environmental Change. J. Exp. Bot. 2015, 66, 3541–3548.
- Niederhuth, C.E.; Schmitz, R.J. Putting DNA Methylation in Context: From Genomes to Gene Expression in Plants. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 149–156.
- Yan, X.; Ma, L.; Pang, H.; Wang, P.; Liu, L.; Cheng, Y.; Cheng, J.; Guo, Y.; Li, Q. Methionine Synthase1 Is Involved in Chromatin Silencing by Maintaining DNA and Histone Methylation. Plant Physiol. 2019, 181, 249–261.
- Zhang, Y.; Harris, C.J.; Liu, Q.; Liu, W.; Ausin, I.; Long, Y.; Xiao, L.; Feng, L.; Chen, X.; Xie, Y.; et al. Large-Scale Comparative Epigenomics Reveals Hierarchical Regulation of Non-CG Methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E1069–E1074.
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for Maintenance of CpXpG Methylation. Science 2001, 292, 2077–2080.
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 2013, 153, 193–205.
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat. Commun. 2019, 10, 729.
- Grimanelli, D.; Ingouff, M. DNA Methylation Readers in Plants. J. Mol. Biol. 2020, 432, 1706–1717.
- Yu, Z.; Zhang, G.; Teixeira da Silva, J.A.; Li, M.; Zhao, C.; He, C.; Si, C.; Zhang, M.; Duan, J. Genome-Wide Identification and Analysis of DNA Methyltransferase and Demethylase Gene Families in Dendrobium officinale Reveal their Potential Functions in Polysaccharide Accumulation. BMC Plant Biol. 2021, 21, 21.
- Gallego-Bartolomé, J. DNA Methylation in Plants: Mechanisms and Tools for Targeted Manipulation. New Phytol. 2020, 227, 38–44.
- Gao, Z.; Liu, H.-L.; Daxinger, L.; Pontes, O.; He, X.; Qian, W.; Lin, H.; Xie, M.; Lorkovic, Z.J.; Zhang, S.; et al. An RNA Polymerase II- and AGO4-Associated Protein Acts in RNA-Directed DNA Methylation. Nature 2010, 465, 106–109.
- Matzke, M.A.; Kanno, T.; Matzke, A.J.M. RNA-Directed DNA Methylation: The Evolution of a Complex Epigenetic Pathway in Flowering Plants. Annu. Rev. Plant Biol. 2015, 66, 243–267.
- Williams, B.P.; Gehring, M. Principles of Epigenetic Homeostasis Shared Between Flowering Plants and Mammals. Trends Genet. 2020, 36, 751–763.
- Beech, E.; Rivers, M.; Oldfield, S.; Smith, P.P. GlobalTreeSearch: The First Complete Global Database of Tree Species and Country Distributions. J. Sustain. For. 2017, 36, 454–489.
- Feng, S.; Jacobsen, S.E. Epigenetic Modifications in Plants: An Evolutionary Perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186.
- Schmitz, R.J.; Ecker, J.R. Epigenetic and Epigenomic Variation in Arabidopsis thaliana. Trends Plant Sci. 2012, 17, 149–154.
- Colomé-Tatché, M.; Cortijo, S.; Wardenaar, R.; Morgado, L.; Lahouze, B.; Sarazin, A.; Etcheverry, M.; Martin, A.; Feng, S.; Duvernois-Berthet, E.; et al. Features of the Arabidopsis Recombination Landscape Resulting from the Combined Loss of Sequence Variation and DNA Methylation. Proc. Natl. Acad. Sci. USA 2012, 109, 16240–16245.
- Ding, Y.; Virlouvet, L.; Liu, N.; Riethoven, J.-J.; Fromm, M.; Avramova, Z. Dehydration Stress Memory Genes of Zea mays; Comparison with Arabidopsis thaliana. BMC Plant. Biol. 2014, 14, 141.
- Yong-Villalobos, L.; González-Morales, S.I.; Wrobel, K.; Gutiérrez-Alanis, D.; Cervantes-Peréz, S.A.; Hayano-Kanashiro, C.; Oropeza-Aburto, A.; Cruz-Ramírez, A.; Martínez, O.; Herrera-Estrella, L. Methylome Analysis Reveals an Important Role for Epigenetic Changes in the Regulation of the Arabidopsis Response to Phosphate Starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E7293–E7302.
- Kawakatsu, T.; Huang, S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166, 492–505.
- Deng, Y.; Zhai, K.; Xie, Z.; Yang, D.; Zhu, X.; Liu, J.; Wang, X.; Qin, P.; Yang, Y.; Zhang, G.; et al. Epigenetic Regulation of Antagonistic Receptors Confers Rice Blast Resistance with Yield Balance. Science 2017, 355, 962–965.
- Bhat, S.S.; Bielewicz, D.; Jarmolowski, A.; Szweykowska-Kulinska, Z. N6-Methyladenosine (M6A): Revisiting the Old with Focus on New, an Arabidopsis thaliana Centered Review. Genes 2018, 9, 596.
- Boquete, M.T.; Muyle, A.; Alonso, C. Plant Epigenetics: Phenotypic and Functional Diversity beyond the DNA Sequence. Am. J. Bot. 2021, 108, 553–558.
- Liu, Q.A. The Impact of Climate Change on Plant Epigenomes. Trends Genet. 2013, 29, 503–505.
- Keller, T.E.; Lasky, J.R.; Yi, S.V. The Multivariate Association between Genomewide DNA Methylation and Climate across the Range of Arabidopsis thaliana. Mol. Ecol. 2016, 25, 1823–1837.
- Sork, V.L. Genomic Studies of Local Adaptation in Natural Plant Populations. J. Hered. 2018, 109, 3–15.
- He, Y.; Li, Z. Epigenetic Environmental Memories in Plants: Establishment, Maintenance, and Reprogramming. Trends Genet. 2018, 34, 856–866.
- Perrone, A.; Martinelli, F. Plant Stress Biology in Epigenomic Era. Plant Sci. 2020, 294, 110376.
- Srikant, T.; Drost, H.-G. How Stress Facilitates Phenotypic Innovation Through Epigenetic Diversity. Front. Plant Sci. 2021, 11, 606800.
- Espinas, N.A.; Saze, H.; Saijo, Y. Epigenetic Control of Defense Signaling and Priming in Plants. Front. Plant Sci. 2016, 7, 1201.
- Boyko, A.; Kovalchuk, I. Epigenetic control of plant stress response. Environ. Mol. Mutagenes. 2008, 49, 61–72.
- Fleta-Soriano, E.; Munné-Bosch, S. Stress Memory and the Inevitable Effects of Drought: A Physiological Perspective. Front. Plant Sci. 2016, 7, 143.
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A Role for Epigenetic Regulation in the Adaptation and Stress Responses of Non-Model Plants. Front. Plant Sci. 2019, 10, 246.
- Pascual, J.; Cañal, M.; Correia, B.; Escandón, M.; Hasbun, R.; Meijón, M.; Pinto, G.; Valledor, L. Can Epigenetics Help Forest Plants to Adapt to Climate Change? In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2014; pp. 125–146.
- Gugger, P.F.; Fitz-Gibbon, S.; PellEgrini, M.; Sork, V.L. Species-Wide Patterns of DNA Methylation Variation in Quercus lobata and their Association with Climate Gradients. Mol. Ecol. 2016, 25, 1665–1680.
- Le Gac, A.-L.; Lafon-Placette, C.; Delaunay, A.; Maury, S. Developmental, Genetic and Environmental Variations of Global DNA Methylation in the First Leaves Emerging from the Shoot Apical Meristem in Poplar Trees. Plant Signal. Behav. 2019, 14, 1596717.
- Jacinto Pereira, W.; de Castro Rodrigues Pappas, M.; Camargo Campoe, O.; Stape, J.L.; Grattapaglia, D.; Joannis Pappas, G., Jr. Patterns of DNA Methylation Changes in Elite Eucalyptus Clones across Contrasting Environments. For. Ecol. Manag. 2020, 474, 118319.
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604.
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway Spruce Genome Sequence and Conifer Genome Evolution. Nature 2013, 497, 579–584.
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb White Spruce (Picea glauca) Genome from Whole-Genome Shotgun Sequencing Data. Bioinformatics 2013, 29, 1492–1497.
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The Genome of Eucalyptus grandis. Nature 2014, 510, 356–362.
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and Assembly of the 22-Gb Loblolly Pine Genome. Genetics 2014, 196, 875–890.
- Gonzalez-Ibeas, D.; Martinez-Garcia, P.J.; Famula, R.A.; Delfino-Mix, A.; Stevens, K.A.; Loopstra, C.A.; Langley, C.H.; Neale, D.B.; Wegrzyn, J.L. Assessing the Gene Content of the Megagenome: Sugar Pine (Pinus lambertiana). G3 Genes Genomes Genet. 2016, 6, 3787–3802.
- Guan, Y.; Li, S.-G.; Fan, X.-F.; Su, Z.-H. Application of Somatic Embryogenesis in Woody Plants. Front. Plant Sci. 2016, 7, 938.
- Sollars, E.S.A.; Harper, A.L.; Kelly, L.J.; Sambles, C.M.; Ramirez-Gonzalez, R.H.; Swarbreck, D.; Kaithakottil, G.; Cooper, E.D.; Uauy, C.; Havlickova, L.; et al. Genome Sequence and Genetic Diversity of European Ash Trees. Nature 2017, 541, 212–216.
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-Fir Genome Sequence Reveals Specialization of the Photosynthetic Apparatus in Pinaceae. G3 Genes Genomes Genet. 2017, 7, 3157–3167.
- Salojärvi, J.; Smolander, O.-P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J.; et al. Genome Sequencing and Population Genomic Analyses Provide Insights into the Adaptive Landscape of Silver Birch. Nat. Genet. 2017, 49, 904–912.
- Bondar, E.I.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Siberian Larch (Larix sibirica Ledeb.) Chloroplast Genome and Development of Polymorphic Chloroplast Markers. BMC Bioinform. 2019, 20, 38.
- Mishra, B.; Gupta, D.K.; Pfenninger, M.; Hickler, T.; Langer, E.; Nam, B.; Paule, J.; Sharma, R.; Ulaszewski, B.; Warmbier, J.; et al. A Reference Genome of the European Beech (Fagus sylvatica L.). GigaScience 2018, 7, giy063.
- Mosca, E.; Cruz, F.; Gómez-Garrido, J.; Bianco, L.; Rellstab, C.; Brodbeck, S.; Csilléry, K.; Fady, B.; Fladung, M.; Fussi, B.; et al. A Reference Genome Sequence for the European Silver Fir (Abies alba Mill.): A Community-Generated Genomic Resource. G3 Genes Genomes Genet. 2019, 9, 2039–2049.
- Wang, W.; Das, A.; Kainer, D.; Schalamun, M.; Morales-Suarez, A.; Schwessinger, B.; Lanfear, R. The Draft Nuclear Genome Assembly of Eucalyptus pauciflora: A Pipeline for Comparing de Novo Assemblies. GigaScience 2020, 9, giz160.
- Plitta-Michalak, B.P.; Naskręt-Barciszewska, M.Z.; Kotlarski, S.; Tomaszewski, D.; Tylkowski, T.; Barciszewski, J.; Chmielarz, P.; Michalak, M. Changes in Genomic 5-Methylcytosine Level Mirror the Response of Orthodox (Acer platanoides L.) and Recalcitrant (Acer pseudoplatanus L.) Seeds to Severe Desiccation. Tree Physiol. 2018, 38, 617–629.
- Raj, S.; Bräutigam, K.; Hamanishi, E.T.; Wilkins, O.; Thomas, B.R.; Schroeder, W.; Mansfield, S.D.; Plant, A.L.; Campbell, M.M. Clone History Shapes Populus Drought Responses. Proc. Natl. Acad. Sci. USA 2011, 108, 12521–12526.
- Lafon-Placette, C.; Le Gac, A.-L.; Chauveau, D.; Segura, V.; Delaunay, A.; Lesage-Descauses, M.-C.; Hummel, I.; Cohen, D.; Jesson, B.; Le Thiec, D.; et al. Changes in the Epigenome and Transcriptome of the Poplar Shoot Apical Meristem in Response to Water Availability Affect Preferentially Hormone Pathways. J. Exp. Bot. 2018, 69, 537–551.
- Fox, H.; Doron-Faigenboim, A.; Kelly, G.; Bourstein, R.; Attia, Z.; Zhou, J.; Moshe, Y.; Moshelion, M.; David-Schwartz, R. Transcriptome Analysis of Pinus Halepensis under Drought Stress and during Recovery. Tree Physiol. 2018, 38, 423–441.
- Li, Q.; Rana, K.; Xiong, Z.; Ge, X.; Li, Z.; Song, H.; Qian, W. Genetic and Epigenetic Alterations in Hybrid and Derived Hexaploids between Brassica napus and B. oleracea Revealed by SSR and MSAP Analysis. Acta Physiol. Plant. 2019, 41, 61.
- Cicatelli, A.; Todeschini, V.; Lingua, G.; Biondi, S.; Torrigiani, P.; Castiglione, S. Epigenetic Control of Heavy Metal Stress Response in Mycorrhizal versus Non-Mycorrhizal Poplar Plants. Environ. Sci. Pollut. Res. 2014, 21, 1723–1737.
- Yakovlev, I.A.; Asante, D.K.A.; Fossdal, C.G.; Junttila, O.; Johnsen, Ø. Differential Gene Expression Related to an Epigenetic Memory Affecting Climatic Adaptation in Norway Spruce. Plant Sci. 2011, 180, 132–139.
- Conde, D.; Gac, A.-L.L.; Perales, M.; Dervinis, C.; Kirst, M.; Maury, S.; González-Melendi, P.; Allona, I. Chilling-Responsive DEMETER-LIKE DNA Demethylase Mediates in Poplar Bud Break. Plant Cell Environ. 2017, 40, 2236–2249.
- Deng, X.; Wang, J.; Li, Y.; Wu, S.; Yang, S.; Chao, J.; Chen, Y.; Zhang, S.; Shi, M.; Tian, W. Comparative Transcriptome Analysis Reveals Phytohormone Signalings, Heat Shock Module and ROS Scavenger Mediate the Cold-Tolerance of Rubber Tree. Sci. Rep. 2018, 8, 4931.
- Wang, B.; Zhang, M.; Fu, R.; Qian, X.; Rong, P.; Zhang, Y.; Jiang, P.; Wang, J.; Lu, X.; Wang, D.; et al. Epigenetic Mechanisms of Salt Tolerance and Heterosis in Upland Cotton (Gossypium hirsutum L.) Revealed by Methylation-Sensitive Amplified Polymorphism Analysis. Euphytica 2016, 208, 477–491.
- Liu, C.; Li, H.; Lin, J.; Wang, Y.; Xu, X.; Cheng, Z.-M.; Chang, Y. Genome-Wide Characterization of DNA Demethylase Genes and Their Association with Salt Response in Pyrus. Genes 2018, 9, 398.
- Liu, J.-G.; Han, X.; Yang, T.; Cui, W.-H.; Wu, A.-M.; Fu, C.-X.; Wang, B.-C.; Liu, L.-J. Genome-Wide Transcriptional Adaptation to Salt Stress in Populus. BMC Plant. Biol. 2019, 19, 367.
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic Variation in Mangrove Plants Occurring in Contrasting Natural Environment. PLoS ONE 2010, 5, e10326.
- Ramirez-Prado, J.S.; Abulfaraj, A.A.; Rayapuram, N.; Benhamed, M.; Hirt, H. Plant Immunity: From Signaling to Epigenetic Control of Defense. Trends Plant Sci. 2018, 23, 833–844.
- Skrøppa, T.; Tollefsrud, M.M.; Sperisen, C.; Johnsen, Ø. Rapid Change in Adaptive Performance from One Generation to the next in Picea abies—Central European Trees in a Nordic Environment. Tree Genet. Genomes 2010, 6, 93–99.
- Yakovlev, I.A.; Lee, Y.; Rotter, B.; Olsen, J.E.; Skrøppa, T.; Johnsen, Ø.; Fossdal, C.G. Temperature-Dependent Differential Transcriptomes during Formation of an Epigenetic Memory in Norway Spruce Embryogenesis. Tree Genet. Genomes 2014, 10, 355–366.
- Yakovlev, I.A.; Carneros, E.; Lee, Y.; Olsen, J.E.; Fossdal, C.G. Transcriptional Profiling of Epigenetic Regulators in Somatic Embryos during Temperature Induced Formation of an Epigenetic Memory in Norway Spruce. Planta 2016, 243, 1237–1249.
- Wang, P.; Xia, H.; Zhang, Y.; Zhao, S.; Zhao, C.; Hou, L.; Li, C.; Li, A.; Ma, C.; Wang, X. Genome-Wide High-Resolution Mapping of DNA Methylation Identifies Epigenetic Variation across Embryo and Endosperm in Maize (Zea may). BMC Genom. 2015, 16, 21.
- Lamelas, L.; Valledor, L.; Escandón, M.; Pinto, G.; Cañal, M.J.; Meijón, M. Integrative Analysis of the Nuclear Proteome in Pinus Radiata Reveals Thermopriming Coupled to Epigenetic Regulation. J. Exp. Bot. 2020, 71, 2040–2057.
- Le Gac, A.-L.; Lafon-Placette, C.; Chauveau, D.; Segura, V.; Delaunay, A.; Fichot, R.; Marron, N.; Le Jan, I.; Berthelot, A.; Bodineau, G.; et al. Winter-Dormant Shoot Apical Meristem in Poplar Trees Shows Environmental Epigenetic Memory. J. Exp. Bot. 2018, 69, 4821–4837.
- Forestan, C.; Farinati, S.; Zambelli, F.; Pavesi, G.; Rossi, V.; Varotto, S. Epigenetic Signatures of Stress Adaptation and Flowering Regulation in Response to Extended Drought and Recovery in Zea mays. Plant. Cell Environ. 2020, 43, 55–75.
- Sow, M.D.; Segura, V.; Chamaillard, S.; Jorge, V.; Delaunay, A.; Lafon-Placette, C.; Fichot, R.; Faivre-Rampant, P.; Villar, M.; Brignolas, F.; et al. Narrow-Sense Heritability and PST Estimates of DNA Methylation in Three Populus nigra L. Populations under Contrasting Water Availability. Tree Genet. Genomes 2018, 14, 78.
- Guarino, F.; Cicatelli, A.; Brundu, G.; Heinze, B.; Castiglione, S. Epigenetic Diversity of Clonal White Poplar (Populus alba L.) Populations: Could Methylation Support the Success of Vegetative Reproduction Strategy? PLoS ONE 2015, 10, e0131480.
- Thellier, M.; Lüttge, U. Plant Memory: A Tentative Model. Plant Biol. 2013, 15, 1–12.
- Rico, L.; Ogaya, R.; Barbeta, A.; Peñuelas, J. Changes in DNA Methylation Fingerprint of Quercus Ilex Trees in Response to Experimental Field Drought Simulating Projected Climate Change. Plant Biol. 2014, 16, 419–427.



