 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carla Di Dedda | + 1432 word(s) | 1432 | 2021-10-22 03:41:29 | | | |
| 2 | Catherine Yang | + 1 word(s) | 1433 | 2021-12-20 09:54:17 | | | | |
| 3 | Catherine Yang | + 1 word(s) | 1433 | 2021-12-20 09:57:40 | | |
Video Upload Options
Glut1 is the main glucose transporter for glucose uptake and is expressed in nearly all mammalian cells. Glut1 is encoded by the Slc2a1 gene, and consists of a sugar-binding pocket facing the outer cell in the outward open conformation. Binding of glucose causes a conformational change so that Glut1 opens into the cytoplasm and release glucose inside the cell. Glut1 exhibit a Michaelis–Menten constant (Km) about 1mM, that is less than the normal blood glucose level (5.5mM), resulting in the continuous transport of glucose inside cells at an essentially constant rate. Similar to the insulin-responsive glucose transporter Glut4, Glut1 cell surface localization is controlled by extrinsic signals. Up-regulation of GLUT1 and consequent increase in glucose uptake occur in some conditions that require extraordinary metabolic needs, such as in the case of tumors and in the case of T cell activation.
1. Novel drugs targeting Glut1
| Name | Structure | MW | IC50 (µ) | Characteristics | Human Cell Target (ref) |
|---|---|---|---|---|---|
| STF-31 | 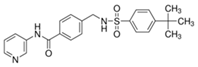 |
423.53 | 1 | Low solubility | Renal cancer RCC4 [2] CD4+ T cells [3] Beta cells [4] |
| WZB-117 | 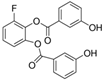 |
368.31 | 0.5 | High solubility | Multiple cancer cell lines [5] Autoreactive CD8+ T cells [6] |
| BAY 876 | 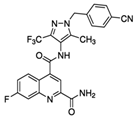 |
496.42 | 0.002 | Highly selective Orally bioavailable | Colon cancer DLD1 [7] |
2. Potential off-Target and Side Effects of Pharmacological Glut1 Blockade
References
- 37 Granchi, C.; Fortunato, S.; Minutolo, F. Anticancer agents interacting with membrane glucose transporters. Medchemcomm 2016, 7, 1716–1729.
- Chan, D.A.; Sutphin, P.D.; Nguyen, P.; Turcotte, S.; Lai, E.W.; Banh, A.; Reynolds, G.E.; Chi, J.T.; Wu, J.; Solow-Cordero, D.E.; et al. Targeting GLUT1 and the Warburg effect in renal cell carcinoma by chemical synthetic lethality. Sci. Transl. Med. 2011, 3, 94ra70.
- Williamson, M.K.; Coombes, N.; Juszczak, F.; Athanasopoulos, M.; Khan, M.B.; Eykyn, T.R.; Srenathan, U.; Taams, L.S.; Zeidler, J.D.; Da Poian, A.T.; et al. Upregulation of glucose uptake and hexokinase activity of primary human CD4+ T cells in response to infection with HIV-1. Viruses 2018, 10, 114.
- Pingitore, A.; Ruz-Maldonado, I.; Liu, B.; Huang, G.C.; Choudhary, P.; Persaud, S.J. Dynamic Profiling of Insulin Secretion and ATP Generation in Isolated Human and Mouse Islets Reveals Differential Glucose Sensitivity. Cell. Physiol. Biochem. 2017, 44, 1352–1359.
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682.
- Vignali, D.; Cantarelli, E.; Bordignon, C.; Canu, A.; Citro, A.; Annoni, A.; Piemonti, L.; Monti, P. Detection and characterization of CD8+ autoreactive memory Stem T cells in patients with type 1 diabetes. Diabetes 2018, 67, 936–945.
- Siebeneicher, H.; Cleve, A.; Rehwinkel, H.; Neuhaus, R.; Heisler, I.; Müller, T.; Bauser, M.; Buchmann, B. Identification and Optimization of the First Highly Selective GLUT1 Inhibitor BAY-876. Chem. Med. Chem. 2016, 11, 2261–2271.
- Ojelabi, O.A.; Lloyd, K.P.; Simon, A.H.; De Zutter, J.K.; Carruthers, A. WZB117 (2-fluoro-6-(m-hydroxybenzoyloxy) Phenyl m-Hydroxybenzoate) inhibits GLUT1-mediated sugar transport by binding reversibly at the exofacial sugar binding site. J. Biol. Chem. 2016, 291, 26762–26772.
- Adams, D.J.; Ito, D.; Rees, M.G.; Seashore-Ludlow, B.; Puyang, X.; Ramos, A.H.; Cheah, J.H.; Clemons, P.A.; Warmuth, M.; Zhu, P.; et al. NAMPT is the cellular target of STF-31-like small-molecule probes. ACS Chem. Biol. 2014, 9, 2247–2254.
- Ma, Y.; Wang, W.; Idowu, M.O.; Oh, U.; Wang, X.Y.; Temkin, S.M.; Fang, X. Ovarian cancer relies on glucose transporter 1 to fuel glycolysis and growth: Anti-tumor activity of BAY-876. Cancers 2019, 11, 1–16.
- Helgerson, A.L.; Carruthers, A. Equilibrium ligand binding to the human erythrocyte sugar transporter. Evidence for two sugar-binding sites per carrier. J. Biol. Chem. 1987, 262, 5464–5475.
- Sebastian, A.; Harris, S.; Ottaway, J.; Todd, K.; Morris, R. Improved mineral balance and skeletal metabolism in postmenopausal women treated with potassium bicarbonate. N. Engl. J. Med. 1994, 330, 1776–1781.
- De Giorgis, V.; Veggiotti, P. GLUT1 Deficiency syndrome 2013: Current state of the art. Seizure 2013, 22, 803–811.
- Pearson, T.S.; Akman, C.; Hinton, V.J.; Engelstad, K.; De Vivo, D.C. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS). Curr. Neurol. Neurosci. Rep. 2013, 13, 1–9.
- Leen, W.G.; Taher, M.; Verbeek, M.M.; Kamsteeg, E.J.; Van De Warrenburg, B.P.; Willemsen, M.A. GLUT1 deficiency syndrome into adulthood: A follow-up study. J. Neurol. 2014, 261, 589–599.
- Raez, L.E.; Papadopoulos, K.; Ricart, A.D.; Chiorean, E.G.; Dipaola, R.S.; Stein, M.N.; Rocha Lima, C.M.; Schlesselman, J.J.; Tolba, K.; Langmuir, V.K.; et al. A phase i dose-escalation trial of 2-deoxy-d-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2013, 71, 523–530.
- De Vos, A.; Heimberg, H.; Quartier, E.; Huypens, P.; Bouwens, L.; Pipeleers, D.; Schuit, F. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J. Clin. Invest. 1995, 96, 2489–2495.
- Kover, K.; Tong, P.Y.; Watkins, D.; Clements, M.; Stehno-Bittel, L.; Novikova, L.; Bittel, D.; Kibiryeva, N.; Stuhlsatz, J.; Yan, Y.; et al. Expression and Regulation of Nampt in Human Islets. PLoS ONE 2013, 8, 1–11.
- Heimberg, H.; De Vos, A.; Pipeleers, D.; Thorens, B.; Schuit, F. Differences in glucose transporter gene expression between rat pancreatic α- and β-cells are correlated to differences in glucose transport but not in glucose utilization. J. Biol. Chem. 1995, 270, 8971–8977.

