 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | mohammad houshmand | + 2166 word(s) | 2166 | 2021-12-13 07:52:58 | | | |
| 2 | Conner Chen | Meta information modification | 2166 | 2021-12-21 08:41:38 | | |
Video Upload Options
Chronic myeloid leukemia stem cells (CML LSCs) are a rare and quiescent population that are resistant to tyrosine kinase inhibitors (TKI). When TKI therapy is discontinued in CML patients in deep, sustained and apparently stable molecular remission, these cells in approximately half of the cases restart to grow, resuming the leukemic process. The elimination of these TKI resistant leukemic stem cells is therefore an essential step in increasing the percentage of those patients who can reach a successful long-term treatment free remission (TFR). The understanding of the biology of the LSCs and the identification of the differences, phenotypic and/or metabolic, that could eventually allow them to be distinguished from the normal hematopoietic stem cells (HSCs) are therefore important steps in designing strategies to target LSCs in a rather selective way, sparing the normal counterparts.
1. WNT Signaling Pathway
2. Hedgehog Signaling Pathway
3. PI3K-AKT Pathway
4. JAK-STAT Pathway
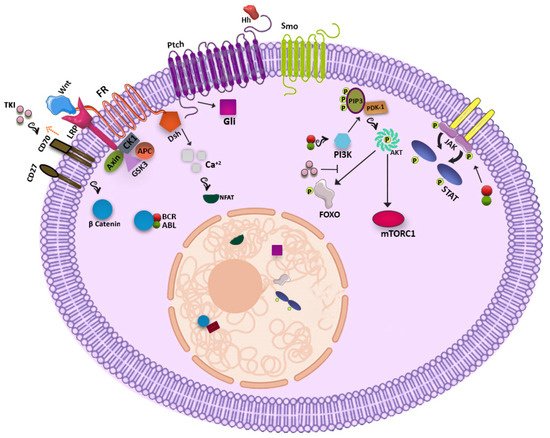
5. Other Players
5.1. Blk
5.2. EZH2
5.3. Fap-1
5.4. HIF-1
5.5. PML
5.6. PP2A
5.7. ALOX5
5.8. SIRT1
| CML Clinical Trial with New Therapeutic Agents (Non-TKIs) | |||||
|---|---|---|---|---|---|
| Generic Name | Brand Name | Clinical Trial Identifier | Target | Start Date | Status |
| Sirolimus | Rapamune | NCT00101088 | mTOR inhibitors | 10-Jan-05 | Terminated |
| Sorafenib | Nexavar | NCT00661180 | Multikinase inhibitors, VEGF/VEGFR inhibitors | 18-Apr-08 | Completed |
| Sunitinib | Sutent | NCT00387426 | Multikinase inhibitors, VEGF/VEGFR inhibitors | 13-Oct-06 | Completed |
| Ruxolitinib | Jakafi | NCT02253277 | JAK/STAT inhibitors | 1-Oct-14 | Completed |
| Axitinib | Inlyta | NCT02782403 | Multikinase inhibitors, VEGF/VEGFR inhibitors | 25-May-16 | Terminated |
| Ibrutinib | Imbruvica | NCT03267186 | BTK inhibitors | 30-Aug-17 | Ongoing |
| Midostaurin | Rydapt | NCT02115295 | Multikinase inhibitors | 16-Apr-14 | Ongoing |
| PRI-724 | - | NCT01606579 | Wnt/β-catenin inhibitors | 25-May-12 | Completed |
| BP1001 | - | NCT02923986 | Grb2 | 5-Oct-16 | Withdrawn |
| Tipifarnib | Zarnestra | NCT00040105 | Farnesyl transferase | 21-Jun-02 | Completed |
| Lonafarnib | SCH66336 | NCT00047502 | Farnesyl transferase | 9-Oct-02 | Completed |
| Rapamycin | Sirolimus | NCT00780104 | mTOR | 27-Oct-08 | Completed |
| RAD001 | Everolimus | NCT01188889 | mTOR | 26-Aug-10 | Withdrawn |
| Panobinostat | LBH589 | NCT00451035 | Histone deacetylase | 22-Mar-07 | Terminated |
| Azacytidine | Vidaza | NCT03895671 | Hypomethylating agents | 29-Mar-19 | Ongoing |
| MK-0457 | Tozasertib | NCT00405054 | Aurora kinase pathway inhibitors | 29-Nov-06 | Terminated |
| Venetoclax | Venclexta | NCT02689440 | BCL-2 inhibitors | 24-Feb-16 | Ongoing |
| Temsirolimus | Torisel | NCT00101088 | mTOR | 10-Jan-05 | Terminated |
| Abemaciclib | Verzenio | NCT03878524 | CDK 4/6 inhibitors | 18-Mar-19 | Ongoing |
| Alemtuzumab | Lemtrada/campath | NCT00626626 | CD52 monoclonal antibodies | 29-Feb-08 | Terminated |
| Bevacizumab | Avastin | NCT00023920 | VEGF/VEGFR inhibitors | 27-Jan-03 | Terminated |
| Blinatumomab | Blincyto | NCT02790515 | Miscellaneous antineoplastic | 6-Jun-16 | Ongoing |
| Ipilimumab | Yervoy | NCT00732186 | Anti-CTLA-4 monoclonal antibodies | 11-Aug-08 | Withdrawn |
| Nivolumab | Opdivo | NCT02011945 | Anti-PD-1 monoclonal antibodies | 16-Dec-13 | Completed |
| Rituximab | Rituxan | NCT03455517 | Antirheumatics, CD20 monoclonal antibodies | 6-Mar-18 | Terminated |
References
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26.
- Zhao, C.; Blum, J.; Chen, A.; Kwon, H.Y.; Jung, S.H.; Cook, J.M.; Lagoo, A.; Reya, T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell 2007, 12, 528–541.
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667.
- Zhang, B.; Li, M.; McDonald, T.; Holyoake, T.L.; Moon, R.T.; Campana, D.; Shultz, L.; Bhatia, R. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood 2013, 121, 1824–1838.
- Liu, N.; Zang, S.; Liu, Y.; Wang, Y.; Li, W.; Liu, Q.; Ji, M.; Ma, D.; Ji, C. FZD7 regulates BMSCs-mediated protection of CML cells. Oncotarget 2016, 7, 6175–6187.
- Zhou, H.; Mak, P.Y.; Mu, H.; Mak, D.H.; Zeng, Z.; Cortes, J.; Liu, Q.; Andreeff, M.; Carter, B.Z. Combined inhibition of beta-catenin and Bcr-Abl synergistically targets tyrosine kinase inhibitor-resistant blast crisis chronic myeloid leukemia blasts and progenitors in vitro and in vivo. Leukemia 2017, 31, 2065–2074.
- Hu, Y.; Chen, Y.; Douglas, L.; Li, S. beta-Catenin is essential for survival of leukemic stem cells insensitive to kinase inhibition in mice with BCR-ABL-induced chronic myeloid leukemia. Leukemia 2009, 23, 109–116.
- Hanna, A.; Shevde, L.A. Hedgehog signaling: Modulation of cancer properies and tumor mircroenvironment. Mol. Cancer 2016, 15, 24.
- Su, W.; Meng, F.; Huang, L.; Zheng, M.; Liu, W.; Sun, H. Sonic hedgehog maintains survival and growth of chronic myeloid leukemia progenitor cells through beta-catenin signaling. Exp. Hematol. 2012, 40, 418–427.
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249.
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98.
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779.
- Cea, M.; Cagnetta, A.; Cirmena, G.; Garuti, A.; Rocco, I.; Palermo, C.; Pierri, I.; Reverberi, D.; Nencioni, A.; Ballestrero, A.; et al. Tracking molecular relapse of chronic myeloid leukemia by measuring Hedgehog signaling status. Leuk. Lymphoma 2013, 54, 342–352.
- Martelli, A.M.; Evangelisti, C.; Chiarini, F.; Grimaldi, C.; Cappellini, A.; Ognibene, A.; McCubrey, J.A. The emerging role of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in normal myelopoiesis and leukemogenesis. Biochim. Biophys. Acta 2010, 1803, 991–1002.
- Pellicano, F.; Scott, M.T.; Helgason, G.V.; Hopcroft, L.E.; Allan, E.K.; Aspinall-O'Dea, M.; Copland, M.; Pierce, A.; Huntly, B.J.; Whetton, A.D.; et al. The antiproliferative activity of kinase inhibitors in chronic myeloid leukemia cells is mediated by FOXO transcription factors. Stem. Cells 2014, 32, 2324–2337.
- Airiau, K.; Mahon, F.X.; Josselin, M.; Jeanneteau, M.; Belloc, F. PI3K/mTOR pathway inhibitors sensitize chronic myeloid leukemia stem cells to nilotinib and restore the response of progenitors to nilotinib in the presence of stem cell factor. Cell Death Dis. 2013, 4, e827.
- Chai, S.K.; Nichols, G.L.; Rothman, P. Constitutive activation of JAKs and STATs in BCR-Abl-expressing cell lines and peripheral blood cells derived from leukemic patients. J. Immunol. 1997, 159, 4720–4728.
- Xie, S.; Wang, Y.; Liu, J.; Sun, T.; Wilson, M.B.; Smithgall, T.E.; Arlinghaus, R.B. Involvement of Jak2 tyrosine phosphorylation in Bcr-Abl transformation. Oncogene 2001, 20, 6188–6195.
- Samanta, A.; Perazzona, B.; Chakraborty, S.; Sun, X.; Modi, H.; Bhatia, R.; Priebe, W.; Arlinghaus, R. Janus kinase 2 regulates Bcr-Abl signaling in chronic myeloid leukemia. Leukemia 2011, 25, 463–472.
- Madapura, H.S.; Nagy, N.; Ujvari, D.; Kallas, T.; Krohnke, M.C.L.; Amu, S.; Bjorkholm, M.; Stenke, L.; Mandal, P.K.; McMurray, J.S.; et al. Interferon gamma is a STAT1-dependent direct inducer of BCL6 expression in imatinib-treated chronic myeloid leukemia cells. Oncogene 2017, 36, 4619–4628.
- Patel, S.B.; Nemkov, T.; Stefanoni, D.; Benavides, G.A.; Bassal, M.A.; Crown, B.L.; Matkins, V.R.; Camacho, V.; Kuznetsova, V.; Hoang, A.T.; et al. Metabolic alterations mediated by STAT3 promotes drug persistence in CML. Leukemia 2021.
- Prost, S.; Relouzat, F.; Spentchian, M.; Ouzegdouh, Y.; Saliba, J.; Massonnet, G.; Beressi, J.P.; Verhoeyen, E.; Raggueneau, V.; Maneglier, B.; et al. Erosion of the chronic myeloid leukaemia stem cell pool by PPARgamma agonists. Nature 2015, 525, 380–383.
- Zhang, H.; Peng, C.; Hu, Y.; Li, H.; Sheng, Z.; Chen, Y.; Sullivan, C.; Cerny, J.; Hutchinson, L.; Higgins, A.; et al. The Blk pathway functions as a tumor suppressor in chronic myeloid leukemia stem cells. Nat. Genet. 2012, 44, 861–871.
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248–1257.
- Huang, W.; Luan, C.H.; Hjort, E.E.; Bei, L.; Mishra, R.; Sakamoto, K.M.; Platanias, L.C.; Eklund, E.A. The role of Fas-associated phosphatase 1 in leukemia stem cell persistence during tyrosine kinase inhibitor treatment of chronic myeloid leukemia. Leukemia 2016, 30, 1502–1509.
- Cheloni, G.; Tanturli, M.; Tusa, I.; Ho DeSouza, N.; Shan, Y.; Gozzini, A.; Mazurier, F.; Rovida, E.; Li, S.; Dello Sbarba, P. Targeting chronic myeloid leukemia stem cells with the hypoxia-inducible factor inhibitor acriflavine. Blood 2017, 130, 655–665.
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1alpha is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607.
- Lallemand-Breitenbach, V.; de The, H. PML nuclear bodies. Cold Spring Harb. Perspect. Biol. 2010, 2, a000661.
- Kakizuka, A.; Miller, W.H., Jr.; Umesono, K.; Warrell, R.P., Jr.; Frankel, S.R.; Murty, V.V.; Dmitrovsky, E.; Evans, R.M. Chromosomal translocation t(15;17) in human acute promyelocytic leukemia fuses RAR alpha with a novel putative transcription factor, PML. Cell 1991, 66, 663–674.
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078.
- Hong, C.S.; Ho, W.; Zhang, C.; Yang, C.; Elder, J.B.; Zhuang, Z. LB100, a small molecule inhibitor of PP2A with potent chemo- and radio-sensitizing potential. Cancer Biol. Ther. 2015, 16, 821–833.
- Neviani, P.; Harb, J.G.; Oaks, J.J.; Santhanam, R.; Walker, C.J.; Ellis, J.J.; Ferenchak, G.; Dorrance, A.M.; Paisie, C.A.; Eiring, A.M.; et al. PP2A-activating drugs selectively eradicate TKI-resistant chronic myeloid leukemic stem cells. J. Clin. Invest. 2013, 123, 4144–4157.
- Lai, D.; Chen, M.; Su, J.; Liu, X.; Rothe, K.; Hu, K.; Forrest, D.L.; Eaves, C.J.; Morin, G.B.; Jiang, X. PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL(+) human leukemia. Sci. Transl. Med. 2018, 10.
- Massoumi, R.; Sjolander, A. The role of leukotriene receptor signaling in inflammation and cancer. Sci. World J. 2007, 7, 1413–1421.
- Chen, Y.; Hu, Y.; Zhang, H.; Peng, C.; Li, S. Loss of the Alox5 gene impairs leukemia stem cells and prevents chronic myeloid leukemia. Nat. Genet. 2009, 41, 783–792.
- Dolinska, M.; Piccini, A.; Wong, W.M.; Gelali, E.; Johansson, A.S.; Klang, J.; Xiao, P.; Yektaei-Karin, E.; Stromberg, U.O.; Mustjoki, S.; et al. Leukotriene signaling via ALOX5 and cysteinyl leukotriene receptor 1 is dispensable for in vitro growth of CD34(+)CD38(-) stem and progenitor cells in chronic myeloid leukemia. Biochem. Biophys. Res. Commun. 2017, 490, 378–384.
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 2011, 9, 11.
- Yuan, H.; Wang, Z.; Li, L.; Zhang, H.; Modi, H.; Horne, D.; Bhatia, R.; Chen, W. Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 2012, 119, 1904–1914.
- Wang, Z.; Yuan, H.; Roth, M.; Stark, J.M.; Bhatia, R.; Chen, W.Y. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene 2013, 32, 589–598.
- Li, L.; Wang, L.; Li, L.; Wang, Z.; Ho, Y.; McDonald, T.; Holyoake, T.L.; Chen, W.; Bhatia, R. Activation of p53 by SIRT1 inhibition enhances elimination of CML leukemia stem cells in combination with imatinib. Cancer Cell 2012, 21, 266–281.


