Chronic myeloid leukemia stem cells (CML LSCs) are a rare and quiescent population that are resistant to tyrosine kinase inhibitors (TKI). When TKI therapy is discontinued in CML patients in deep, sustained and apparently stable molecular remission, these cells in approximately half of the cases restart to grow, resuming the leukemic process. The elimination of these TKI resistant leukemic stem cells is therefore an essential step in increasing the percentage of those patients who can reach a successful long-term treatment free remission (TFR). The understanding of the biology of the LSCs and the identification of the differences, phenotypic and/or metabolic, that could eventually allow them to be distinguished from the normal hematopoietic stem cells (HSCs) are therefore important steps in designing strategies to target LSCs in a rather selective way, sparing the normal counterparts.
1. WNT Signaling Pathway
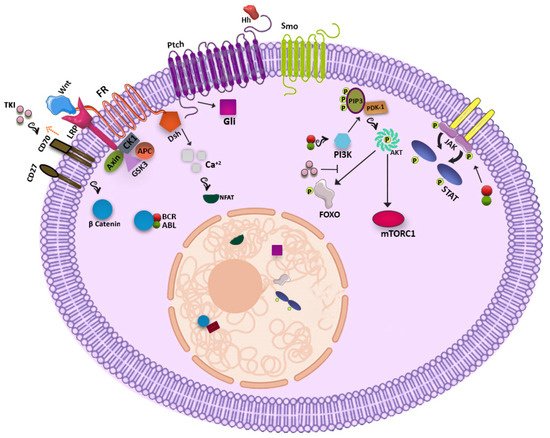
The WNT signaling pathway has been shown to have a significant role in the development of several organs including the hematopoietic system while perturbation of this crucial pathway sparks induction of various types of cancers. In the resting condition, the WNT-β catenin network forms a destruction complex including AXIN, adenomatous polyposis coli (APC), Casein kinase 1 (CK1) and Glycogen synthase kinase 3 (GSK3) and links to β-catenin, providing a binding site for the ubiquitin ligase that leads to β catenin degradation in the proteasome. By contrast, following attachment of WNT to the frizzled receptor (FR), GSK3, CK1, and AXIN bind Lipoprotein receptor-related proteins (LRP) and leave β-catenin free for nucleus localization where it interacts with transcription factors of the TCF/LEF family and promotes gene expression. It has been demonstrated that β-catenin is of paramount importance for self-renewal and long-term maintenance of both HSCs and LSCs [
3]. After induction of BCR-ABL in β-catenin null mice, defect in self-renewal and in engraftment potential of CML LSCs has been observed and this shows that this pathway is essential for normal and leukemic stem cells survival [
4]. It has been shown that BCR-ABL has direct contact with β-catenin and mediates the nucleus transition by making this protein more stable. Furthermore, expression of β-catenin increases with CML progression and is responsible for the increased self-renewal of CML progenitors in blast crisis [
5]. In the bone marrow niche, mesenchymal cells may interact with CML LSCs through the WNT/β- catenin pathway enhancing their proliferation. Therefore, increased expression of β-catenin can be seen as a form of resistance of LSCs allowing their survival [
6,
7] and a combination of WNT/β-catenin inhibitors and TKI could potentially help to get rid of CML LSCs. However, unfortunately this approach has been demonstrated to be too toxic also for normal HSCs as well as for the stem cells of other organs and, at least for the moment, has been abandoned [
8,
9].
2. Hedgehog Signaling Pathway
The Hedgehog signaling pathway which is essential for hematopoiesis, is deregulated in several solid tumors. It is initiated through binding of hedgehog (Hh) ligands (Sonic Hh, Indian Hh, and Desert Hh) to a seven transmembrane receptor called Patched (Ptch). After the consequent activation of the Smoothened protein (Smo) by Ptch, Glioma-Associated Oncogene Homolog (Gli) family transcription factors are activated, which are able to transcribe target genes such as Gli1, Ptch1, bcl2, Cyclin D, and MYC [
10]. In CML high mRNA expression of Hedgehog cascade related proteins underlines the role of this pathway in driving leukemogenesis [
11]. ABL kinase is not needed for the salvation of this cascade [
12]. Whereas studies suggest that Smo targeting does not affect the engraftment potential and the fate decision of normal HSCs, it has been shown that it may potentially reduce the engraftment ability and the colony formation of CML LSCs. Therefore, hitting this pathway should selectively affect LSCs but not normal HSCs [
12,
13]. Indeed, exposure of CML cells containing both wild type BCR-ABL and BCR-ABL1 mutated T315I cells to an Smo inhibitor led to the purging of the mutated clone [
14]. Another study demonstrated that the use of Hedgehog inhibitors not only propels CML LSCs into cycling condition but also restores their susceptibility to TKI [
13,
15]. Again, however clinical trials testing the usefulness of this approach have shown that Hh pathway inhibitors are too toxic and have been finally abandoned.
3. PI3K-AKT Pathway
The known participation of the Phosphatidylinositol-3-kinase (PI3K) signaling pathway in the maintenance and function of normal HSCs drove the attention on the possible role of this cascade in the LSC population. By phosphorylation of phosphatidylinositol (3,4)-bisphosphate (PIP2) by PI3K and formation of phosphatidylinositol-3,4,5-trisphosphate (PIP3), Pyruvate Dehydrogenase Kinase 1 (PDK1) is recruited and associated to PIP3 and phosphorylates AKT, that subsequently activates mTORC1 and phosphorylates the Forkhead box O (FOXO) transcription factors family [
16]. It has been shown that this signaling pathway is activated by BCR-ABL1 and so, in the Ph-positive cell population, it may be more specific with respect to the WNT and Hedgehog signaling pathways. When the BCR-ABL TK activity is on, AKT phosphorylates FOXO transcription factors and does not allow their shift to the nucleus, but TKIs blocking BCR-ABL1 TK activity can promote FOXO nucleus relocalization, restoring their transcriptional activity. Expression of BCL6, that is considered essential for the survival of CML stem cells, and also ATM and CDKN1C, is enhanced by the FOXO transcriptional activity [
16,
17]. Inhibition of mTORC1 did not show an evident effect on CML LSCs, but inhibition of PI3K can restore the vulnerability of CML LSCs to TKIs [
18].
4. JAK-STAT Pathway
In addition, the JAK-STAT pathway plays an important role in CML, but in a strong association with BCR-ABL1 kinase activity. Indeed, STAT1, STAT3, and STAT5 can be activated by BCR-ABL1 directly or indirectly through JAK2 induction and activation by BCR-ABL1. JAK2 activation can be also stimulated by growth factors produced by the mesenchymal cells of the hematopoietic bone marrow niche [
19,
20]. It has been shown that inhibition of the JAK2 by ruxolitinib may reduce the level of BCR-ABL1 protein and may help to overcome resistance [
21]. It was also seen that the combination of Imatinib + INFγ is able to decrease the phosphorylation of STAT5, but it increases the phosphorylation of STAT1, an up-regulator of the survival hint induced by BCL6, clearly delineating another potential pitfall of imatinib and of TKI therapy in general [
22]. In line with these concepts, the use of ruxolitinib (a JAKs inhibitor) together with nilotinib fruitfully in vitro showed activity in suppressing the CML LSC population, without affecting the HSCs. However clinical trials testing this strategy were unsuccessful. It has also been reported that STAT3 dysregulates CML LSC metabolism, and its inhibition may eradicate these resistant cells [
23]. Meanwhile, glitazones an antidiabetic drug by activating peroxisome proliferator-activated receptor-γ (PPARγ) reduces expression of STAT5 that might lead to the elimination of the CML LSC population [
24].
Possible signaling pathways are shown in Figure 1.
Figure 1. Possible signaling pathways in CML LSCs.
5. Other Players
5.1. Blk
It has been shown that Blk as a tyrosine kinase protein has a diminished expression in CML LSCs in contrast to normal cells. Although Blk is regarded as a tumor suppressor, in CML LSCs, via upregulation of p27, BCR-ABL1 downsizes the expression of this protein by modulation of c-myc and Pax5. Meanwhile, overexpression of Blk in CML LSCs inhibits the self-renewal and increases the apoptosis rate, while Blk knock-down does not interfere with the regular HSC activity [
25].
5.2. EZH2
EZH2 as part of the PRC2 complex mediates repression of various genes by trimethylation of histone H3 (H3K27me3). Amplification of EZH2 in CML LSCs and reduction after TKI therapy shows its engagement in the pathogenesis of CML and its dependency on BCR-ABL1 TK activity. It has also been reported that EZH2 inhibitor increases the possibility of CML LSC eradication while sparing the normal HSCs. This effect is enhanced by the combination of TKI and EZH2 Inhibitor [
26].
5.3. Fap-1
Another pathway associated with resistance to elimination is the enhanced expression of Fap-1 in CML LSCs. Fap-1 with its phosphatase activity blocks Fas mediated apoptosis and also stabilizes β-catenin by targeting its inhibitor Gsk3β. Fap-1 activity is accompanied by persistence of CML stem cells and Fap-1 inhibition promotes TKI response and hampers the progression of leukemic cells [
27].
5.4. HIF-1
Hypoxia inducible factor (HIF), constituted of α and β subunits, increases when oxygen concentration is low in order to facilitate the cell adaptation to this new environment. HIF-1 as a transcription factor has a crucial role in regulating survival, proliferation, and maintenance of CML LSCs. Cheloni et al., posited that using acriflavine, an HIF-1 inhibitor, significantly affects the fate of CML cells by c-MYC down regulation and decrease of stemness genes like NANOG, SOX2, and OCT4. As CML LSCs are more dependent on HIF-1 than normal HSCs, the combination of an HIF-1 inhibitor with TKI could represent a new form of strategy to target resistant CML LSCs that reside in the hypoxic region [
28,
29].
5.5. PML
The promyelocyte leukemia protein (PML), by attending in the formation of PML-nuclear bodies (PML-NBs), acts as a tumor suppressor and transcription factor and plays a pivotal role in apoptosis and senescence of normal cells [
30]. The PML gene is already well known because of its involvement in the t(15;17) translocation that causes the fusion of PML with retinoic acid receptor alpha (RAR-alpha) in acute promyelocyte leukemia (APL), determining a differentiation arrest [
31]. Besides, up-regulation of PML in CML LSCs may hamper the cycling of these cells and cause a decrease in their sensitivity to TKIs. It has been shown that targeting PML in CML cells by arsenic trioxide (As
2O
3) leads to PML degradation and triggers cycling of these quiescent cells. This strategy may promote the exhaustion of the CML LSCs restoring their sensitivity to the TKIs [
32].
5.6. PP2A
Protein phosphatase 2 A (PP2A), a serine/threonine phosphatase which is composed by the scaffold (A), regulatory (B), and catalytic (C) subunits, has a role in directing β-catenin pathway, programmed cell death and cell cycle progression [
33]. Until now our knowledge about the role of PP2A in CML LSCs has been limited to its tumor inhibitory effect. In CML this protein is regulated by SET protein activity, and enhancement of SET during the progression of CML from chronic to more advanced phases of the disease determines the downregulation of PP2A. It has been shown that in the LSC population, PP2A reduction provides a stimulus for self-renewal of leukemic cells. Restoring its activity could therefore be useful to decrease the LSC pool [
34]. Various isoforms of PP2A, however are present and while some of them play a suppressive role in many cancers, other isoforms can act differently [
33]. Recently, however, Lai et al. demonstrated that inhibition of PP2A and TKI may efficiently suppress CML LSCs [
35].
5.7. ALOX5
ALOX5 encodes 5-lypoxygenase (5-LO) that converts arachidonic acid into leukotrienes and is involved in inflammatory condition and cancer development [
36]. Targeting ALOX5 hampers the differentiation, the function, and the survival of CML LSCs, while normal HSCs remain uninfluenced. Zileuton (5-LO inhibitor) impairs CML LSC development [
37], but although the oncogenicity of ALOX5 in the mouse model seemed to be compelling, in CML patients it has a low expression and the use of a 5-LO inhibitor does not show particular consequences [
38].
5.8. SIRT1
Sirtuin 1 (SIRT1) is a histone deacetylase that regulates gene expression, metabolic activity and aging within cells [
39]. SIRT1 overexpression in primary CML cells deacetylates many transcription factors including P53, Ku70, and FOX01. This genetic modification promotes drug resistance, survival, and propagation of the leukemic fraction [
40,
41]. SIRT1 targeting in CML LSCs, both by inhibition or knock-down, enhances acetylation of P53 which gives rise to apoptosis and reduction of their growth [
42]. So, applying the combination of TKI and SIRT1 inhibitor maybe a potential approach to tackle leukemogenesis.
Considering the role of different molecules in supporting CML LSC survival and proliferation, many clinical trials have been designed to target these players and are summarized in Table 1.
Table 1. Clinical trials that used non-TKI agents for the treatment of CML.
| CML Clinical Trial with New Therapeutic Agents (Non-TKIs) |
| Generic Name |
Brand Name |
Clinical Trial Identifier |
Target |
Start Date |
Status |
| Sirolimus |
Rapamune |
NCT00101088 |
mTOR inhibitors |
10-Jan-05 |
Terminated |
| Sorafenib |
Nexavar |
NCT00661180 |
Multikinase inhibitors, VEGF/VEGFR inhibitors |
18-Apr-08 |
Completed |
| Sunitinib |
Sutent |
NCT00387426 |
Multikinase inhibitors, VEGF/VEGFR inhibitors |
13-Oct-06 |
Completed |
| Ruxolitinib |
Jakafi |
NCT02253277 |
JAK/STAT inhibitors |
1-Oct-14 |
Completed |
| Axitinib |
Inlyta |
NCT02782403 |
Multikinase inhibitors, VEGF/VEGFR inhibitors |
25-May-16 |
Terminated |
| Ibrutinib |
Imbruvica |
NCT03267186 |
BTK inhibitors |
30-Aug-17 |
Ongoing |
| Midostaurin |
Rydapt |
NCT02115295 |
Multikinase inhibitors |
16-Apr-14 |
Ongoing |
| PRI-724 |
- |
NCT01606579 |
Wnt/β-catenin inhibitors |
25-May-12 |
Completed |
| BP1001 |
- |
NCT02923986 |
Grb2 |
5-Oct-16 |
Withdrawn |
| Tipifarnib |
Zarnestra |
NCT00040105 |
Farnesyl transferase |
21-Jun-02 |
Completed |
| Lonafarnib |
SCH66336 |
NCT00047502 |
Farnesyl transferase |
9-Oct-02 |
Completed |
| Rapamycin |
Sirolimus |
NCT00780104 |
mTOR |
27-Oct-08 |
Completed |
| RAD001 |
Everolimus |
NCT01188889 |
mTOR |
26-Aug-10 |
Withdrawn |
| Panobinostat |
LBH589 |
NCT00451035 |
Histone deacetylase |
22-Mar-07 |
Terminated |
| Azacytidine |
Vidaza |
NCT03895671 |
Hypomethylating agents |
29-Mar-19 |
Ongoing |
| MK-0457 |
Tozasertib |
NCT00405054 |
Aurora kinase pathway inhibitors |
29-Nov-06 |
Terminated |
| Venetoclax |
Venclexta |
NCT02689440 |
BCL-2 inhibitors |
24-Feb-16 |
Ongoing |
| Temsirolimus |
Torisel |
NCT00101088 |
mTOR |
10-Jan-05 |
Terminated |
| Abemaciclib |
Verzenio |
NCT03878524 |
CDK 4/6 inhibitors |
18-Mar-19 |
Ongoing |
| Alemtuzumab |
Lemtrada/campath |
NCT00626626 |
CD52 monoclonal antibodies |
29-Feb-08 |
Terminated |
| Bevacizumab |
Avastin |
NCT00023920 |
VEGF/VEGFR inhibitors |
27-Jan-03 |
Terminated |
| Blinatumomab |
Blincyto |
NCT02790515 |
Miscellaneous antineoplastic |
6-Jun-16 |
Ongoing |
| Ipilimumab |
Yervoy |
NCT00732186 |
Anti-CTLA-4 monoclonal antibodies |
11-Aug-08 |
Withdrawn |
| Nivolumab |
Opdivo |
NCT02011945 |
Anti-PD-1 monoclonal antibodies |
16-Dec-13 |
Completed |
| Rituximab |
Rituxan |
NCT03455517 |
Antirheumatics, CD20 monoclonal antibodies |
6-Mar-18 |
Terminated |
This entry is adapted from the peer-reviewed paper 10.3390/jcm10245805