Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jean-Christophe Cintrat | + 1747 word(s) | 1747 | 2021-12-17 07:17:30 | | | |
| 2 | Amina Yu | Meta information modification | 1747 | 2021-12-17 09:28:07 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Cintrat, J. Pyrrolotriazinone. Encyclopedia. Available online: https://encyclopedia.pub/entry/17248 (accessed on 10 July 2026).
Cintrat J. Pyrrolotriazinone. Encyclopedia. Available at: https://encyclopedia.pub/entry/17248. Accessed July 10, 2026.
Cintrat, Jean-Christophe. "Pyrrolotriazinone" Encyclopedia, https://encyclopedia.pub/entry/17248 (accessed July 10, 2026).
Cintrat, J. (2021, December 17). Pyrrolotriazinone. In Encyclopedia. https://encyclopedia.pub/entry/17248
Cintrat, Jean-Christophe. "Pyrrolotriazinone." Encyclopedia. Web. 17 December, 2021.
Copy Citation
Pyrrolotriazinones, a class of azolotriazinone, are indeed a fused bicyclic compound. Pyrrolotriazinones share common properties with all nitrogen-containing heterocycles.
pyrrolotriazinone
heterocycle
inhibitor
antagonist
1. Pyrrolo[1,2-d][1,2,4]triazin-4(3H)-one (A)
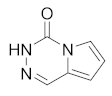
Invasive fungal infections are one of the main concerns in immunocompromised patients. While azoles are the first line therapy in treating candidiasis, side effects such as liver toxicity and appearance of resistance highlight the need for new drugs. In this context, compound 1 (Figure 2), a triazole derivative bearing a pyrrolotriazinone scaffold, has shown broad in vitro antifungal activity [1]. The main biological activity may be due to the azole ring, but one cannot exclude the pharmacophoric influence of the pyrrolotriazinone ring in the inhibition of CYP51. In particular, compound 1 is active in vitro against pathogenic Candida spp. including fluconazole resistant strains as well as Candida albicans (MIC < 0.01 μg mL−1). In addition, the same compound is also active against some filamentous fungi such as Aspergillus fumigatus. Finally, the title compound also demonstrates promising in vivo activity (15 mg/kg) in murine models of lethal systemic infections caused by Candida albicans (Figure 2).
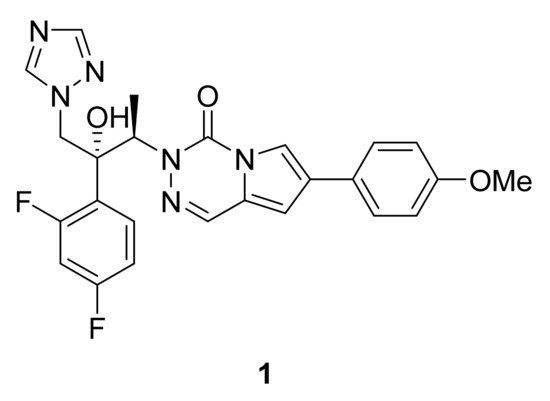
Figure 2. Compound 1, antifungal agent.
2. Pyrrolo[2,1-f][1,2,4]triazin-4(3H)-one (C)
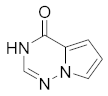
2.1. CRF1 Receptor Antagonists
Corticotropin-releasing factor (CRF), also known as corticotropin-releasing hormone (CRH), is a peptide hormone that plays a key role in the regulation of the hypothalamic-pituitary-adrenal (HPA) axis, with the secretion of two hormones, ACTH, produced by the pituitary gland, and glucocorticoids, produced by the adrenal gland, coordinating endocrine, behavioral and autonomic responses to stress. Indeed, these hormones are increased during stress and are responsible for well-known symptoms, such as stomachaches, nausea, confusion, fatigue, etc. However, in the long term, the damages caused can be much more important, with endocrine (diabetes, hyperthyroidism, hormone deregulation), metabolic, and cardiovascular, as well as psychic, disorders. Although many treatments are already available on the market, the discovery of new potential targets and new treatments remains essential.
Preclinical and clinical studies have suggested that CRF1 antagonists could be promising in the treatment of stress-related disorders. Some studies have also shown the positive impact of these antagonists on intestinal disorders such as irritable bowel syndrome [2][3].
Within this context, and through numerous investigations of structure–activity relationships, Saito et al. [4] have designed a series of pyrrolotriazinone (pyrrolo[2,1-f]triazin-4(3H)-ones) analogues by optimizing binding affinity, antagonistic activity and stability. Compound 2 (Figure 3) was the most interesting, showing very good binding affinity (IC50: 5.3 nM) and antagonistic activity on the CRF1 receptor (EC50: 3.2 nM). It has also been tested for its effect on anxiety using rat elevated plus maze test, showing efficacy in a dose–response manner.
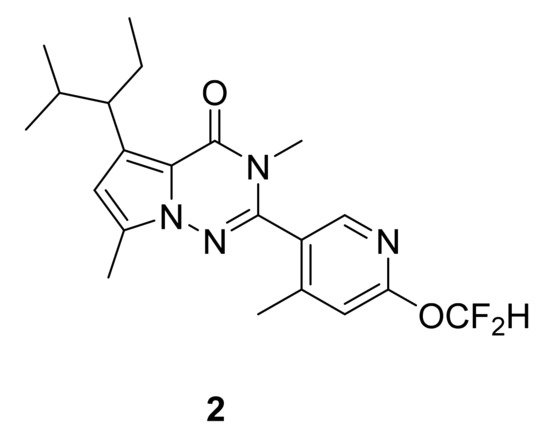
Figure 3. Compound 2, CRF1 receptor antagonist.
2.2. MCHR1 Antagonists
Melanin-concentrating hormone is a hypothalamic neuropeptide and is involved in the neural regulation of food intake and energy homeostasis.
Indeed, acute intracerebroventricular injections of MCH have shown a significant increase in food consumption, and its chronic administration significantly increased food intake, body weight, white adipose tissue mass and liver mass. Moreover, mice that lacked the pro-hormone became hypophagic and lean, and in contrast, mice overexpressing MCH became obese [5]. MCH is also thought to have other roles, notably in stress, anxiety and depression. However, it has so far only been envisioned as a target in the discovery of drugs against obesity.
Devasthale et al. [6] have designed a series of pyrrolotriazinones that were assayed for their antagonistic activity, and the reduction in weight gain in a rat model after 4 days of once-daily oral treatment. Among the tested compounds that exhibited the best brain concentrations, compound 3 (Figure 4) showed the best results, with an MCHR1 inhibition constant (Ki) of 2.3 nM (human) and 1.8 nM (rat) and an impressive 6.9% weight reduction in rat model relative to vehicle after 4 days.
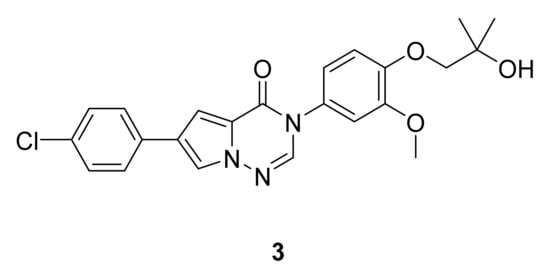
Figure 4. Compound 3, MCHR1 antagonist.
2.3. EP3 Receptor Antagonists
EP3, also known as the prostaglandin receptor EP3, is a receptor for prostaglandin E2 (PGE2) and other prostanoids whose binding induces numerous and diverse cellular responses both physiological and pathological. One can mention the stimulation of digestive secretion; the regulation of blood pressure; the inflammation mechanism, due to the presence of these receptors on platelets [7][8]; the increase of arterial thrombosis, particularly at the level of atherosclerotic vessel walls via their activation and aggregation; and, therefore, an increase in the risk of myocardial infarction and stroke.
In addition, several teams have shown the increased concentration of PGE2 in atherosclerotic vascular walls; therefore, targeting EP3 receptors could reduce thrombosis specifically in atherosclerotic vessels without increasing the risk of bleeding in healthy vessels, which is the main issue with current antithrombotic treatments [9][10][11].
Candish et al. [12] have synthetized a series of pyrrolotriazinone carboxamide and evaluated their EP3 antagonistic ability. Many compounds showed excellent activity, including compounds 4 and 5 yielding an IC50 of 0.3 nM (Figure 5). These compounds’ efficacy was also tested in vivo on mice with laser injury-induced thrombi in atherosclerotic micro vessels.
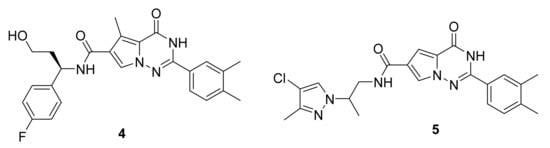
Figure 5. Compound 4 and 5, EP3 receptor antagonists.
2.4. PI3K Inhibitor
Phosphoinositide 3-kinases (PI3Ks), also called phosphatidylinositol 3-kinases, are a family of lipid kinases. They are divided into class I, class II, and class III. Class I is the most extensively studied and the focus of numerous drug discovery efforts. These PI3Ks are activated by cell surface receptors to produce phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 is a key cellular mediator that activates downstream signaling pathways involved in relevant cellular processes such as growth, proliferation, chemotaxis, metabolism, and survival.
Class I PI3K is further subdivided into four different isoforms: PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ. The first two isoforms are ubiquitously expressed and play key roles in cell growth, survival, and proliferation; thus, inhibition of PI3Kα and PI3Kβ is primarily aimed in cancer therapy [13][14]. In the other part, PI3Kγ is widely expressed in granulocytes, monocytes and macrophages, whereas the PI3Kδ isoform is also found in B and T cells. They play a role in the production of cytokines induced by T cell receptors, the degranulation of basophils and mast cells, and the oxidative explosion of neutrophils. Thus, PI3Kδ or PI3Kγ isoform-specific inhibitors may have therapeutic benefits in auto-immune diseases, lymphoma, leukemia, and some inflammatory diseases.
Erra et al. [15][16] have studied PI3Kδ inhibition by the addition of pyrimidine and purine derivatives to the pyrrolotriazinone scaffold and, through an extensive structure–activity relationship study, shown that the compounds had a propeller-shaped conformation where the pyrrolotriazinone moiety was sandwiched into the PI3Kδ hydrophobic binding pocket between Trp760 and Met752. They observed that orally administered compound 6 (LAS191954) has good inhibitory potency (Figure 6), with a PI3Kδ IC50 value of 2.6 nM and very low in vitro metabolism, and is thus indicated for the treatment of inflammatory diseases. They also identified compound 7 (LAS195319), which is administered by inhalation with a PI3Kδ IC50 of 0.5 nM for the treatment of respiratory diseases with an inflammatory component. Jia et al. [17] have attempted to discover and optimize potent and highly selective PI3Kγ−PI3Kδ dual inhibitors (compound 8, PI3Kγ IC50: 4 nM and PI3Kδ IC50: 5 nM, Figure 6). A docking model of the binding site of PI3Kγ showed a similar hydrophobic pocket where the pyrrolotriazinone moiety is sandwiched, formed by Met804, Ile963, and Met953. However, no biological activity study has been reported by Jia’s group, but a higher activity with this double inhibition than with the single inhibition of PI3Kδ is expected.
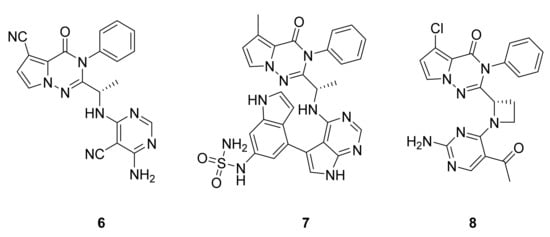
Figure 6. Compounds 6–8, PI3K inhibitors.
2.5. Eg5 Inhibitor
Eg5, or kinesin-5, is an essential molecular motor protein in the mechanism of mitosis. Indeed, its presence allows centrosome separation, which is essential for bipolar spindle formation and sister chromatid segregation. Overexpression of Eg5 can lead to the development of spindle defects and thus to genetic instability and tumors.
On the other hand, its inhibition induces the formation of an aberrant mitotic spindle and then the arrest of the cell cycle at M phase, which leads to apoptosis of the cells and thus to the inhibition of cell proliferation It is therefore conceivable to use it in the treatment of certain human cancers [18]. Furthermore, due to a generically low expression of Eg5 in non-cancerous tissues, a therapy targeting Eg5 will be significantly less toxic compared to current anti-mitotic therapies.
Kinesin-5 motors are phosphorylated at multiple sites within the protein, and this phosphorylation impacts motor activity, localization, and function in mitosis. Thus, Kim et al. [19] have performed a structure–activity relationship study and synthesized compound 9, which showed potent inhibition of Eg5 ATPase (IC50 of ATPase activity ~60 nM) and in vivo efficacy in the P388 murine leukemia model. An X-ray crystal structure of the Eg5 binding site showed a hydrophobic pocket formed by Ile136, Leu214, Phe239, and Leu160 of the protein that allows van der Waals interaction with the pyrrolotriazinone moiety, and hydrophobic interactions between the phenyl and cyclopropyl groups of 9 (Figure 7) with Trp127, Tyr211, Pro137, and Arg119 of the protein.
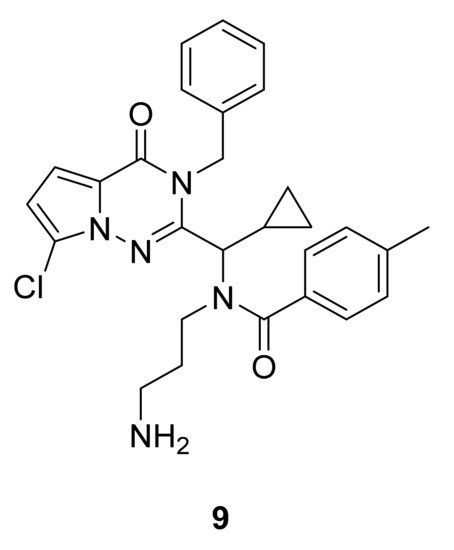
Figure 7. Compound 9, Eg5 inhibitor.
2.6. Sepiapterin Reductase Inhibitors
Sepiapterin reductase plays an important role in the biosynthesis of tetrahydrobiopterin (BH4), which is a key cofactor for a set of enzymes, including nitric oxide synthases (NOSs), aromatic amino acid hydroxylases, and alkylglycerol monooxygenase. Consequently, BH4 is associated with various physiological and pathological biological processes, including monoamine neurotransmitter formation, immune response, cardiovascular function, endothelial dysfunction, and cancer.
Moreover, increased levels of BH4 in injured sensory neurons and inflamed tissues correlate with pain intensity, and its reduction by treatment with RPD inhibitors results in a decrease in pain and inflammation [20]. In a context where therapeutic options for chronic pain are scarce and almost limited to opioids, the discovery and development of new targets is relevant
Tebbe et al. [21] have synthesized a series of pyrrolotriazinone derivatives and screened for their inhibitory activity on SPR. Many of these derivatives have shown very good activity, including compounds 10 and 11 (IC50: 1 nM for both compounds, Figure 8), and significantly reduce pain in behavioral pharmacology models for pain in rats.
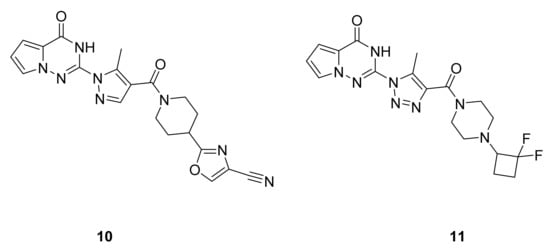
Figure 8. Compounds 10 and 11, sepiapterin reductase inhibitors.
2.7. Ubiquitin-Specific Protease 7 Inhibitors
Ubiquitin-specific protease 7, USP7, is a de-ubiquitin enzyme that catalyzes the removal of ubiquitin from specific target proteins. Given the important role of ubiquitination of proteins, in particular for their degradation and thus their regulation, one can understand the interest in developing inhibitors for these enzymes to treat various pathologies. The involvement of USP7 in the modulation of the p53 signaling pathway has led to an increase in USP7 inhibitors, potential candidates for the treatment of cancers and immunological disorders [22].
Ioannidis et al. [23] have reported a series of pyrrolotriazinones and imidazotriazinones and tested their inhibitory activities against USP7. Among the synthesized compounds, 12 and 13 were the most active, with IC50 values below 200 nM (Figure 9).
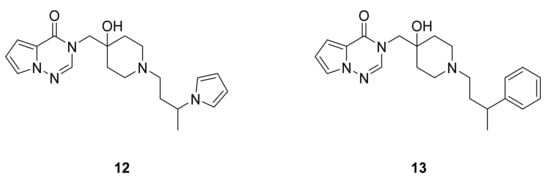
Figure 9. Compounds 12 and 13, USP7 inhibitors.
References
- Montoir, D.; Guillon, R.; Gazzola, S.; Ourliac-Garnier, I.; Soklou, K.E.; Tonnerre, A.; Picot, C.; Planchat, A.; Pagniez, F.; Le Pape, P.; et al. New azole antifungals with a fused triazinone scaffold. Eur. J. Med. Chem. 2020, 189, 112082.
- Tache, Y. Corticotropin releasing factor receptor antagonists: Potential future therapy in gastroenterology? Gut 2004, 53, 919–921.
- Taché, Y.; Kiank, C.; Stengel, A. A role for corticotropin-releasing factor in functional gastrointestinal disorders. Curr. Gastroenterol. Rep. 2009, 11, 270–277.
- Saito, T.; Obitsu, T.; Kohno, H.; Sugimoto, I.; Matsushita, T.; Nishiyama, T.; Hirota, T.; Takeda, H.; Matsumura, N.; Ueno, S.; et al. Pyrrolopyridazines, pyrrolotriazin-4(3H)-ones, and related compounds as novel corticotropin-releasing factor 1 (CRF1) receptor antagonists. Bioorg. Med. Chem. 2012, 20, 1122–1138.
- Pissios, P.; Trombl, D.J.; Tzameli, I.; Maratos-Flier, E. Melanin-concentrating hormone receptor 1 activates extracellular signal-regulated kinase and synergizes with Gs-coupled pathways. Endocrinology 2003, 144, 3514–3523.
- Devasthale, P.; Wang, W.; Mignone, J.; Renduchintala, K.; Radhakrishnan, S.; Dhanapal, J.; Selvaraj, J.; Kuppusamy, R.; Pelleymounter, M.A.; Longhi, D.; et al. Non-basic azolotriazinone MCHR1 antagonists for the treatment of obesity: An empirical brain-exposures-driven candidate selection for in vivo efficacy studies. Bioorg. Med. Chem. Lett. 2015, 25, 4412–4418.
- Paul, B.Z.; Ashby, B.; Sheth, S.B. Distribution of prostaglandin IP and EP receptor subtypes and isoforms in platelets and human umbilical artery smooth muscle cells. Br. J. Haematol. 1998, 102, 1204–1211.
- Cipollone, F.; Prontera, C.; Pini, B.; Marini, M.; Fazia, M.; De Cesare, D.; Iezzi, A.; Ucchino, S.; Boccoli, G.; Saba, V.; et al. Overexpression of Functionally Coupled Cyclooxygenase-2 and Prostaglandin E Synthase in Symptomat-ic Atherosclerotic Plaques as a Basis of Prostaglandin E 2 -Dependent Plaque Instability. Circulation 2001, 104, 921–927.
- Gross, S.; Tilly, P.; Hentsch, D.; Vonesch, J.-L.; Fabre, J.-E. Vascular wall–produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J. Exp. Med. 2007, 204, 311–320.
- Singh, J.; Zeller, W.; Zhou, N.; Hategen, G.; Mishra, R.; Polozov, A.; Yu, P.; Onua, E.; Zhang, J.; Zembower, D.; et al. Antagonists of the EP3 Receptor for Prostaglandin E2 Are Novel Antiplatelet Agents That Do Not Prolong Bleeding. ACS Chem. Biol. 2009, 4, 115–126.
- Fox, S.C.; May, J.A.; Johnson, A.; Hermann, D.; Strieter, D.; Hartman, D.; Heptinstall, S. Effects on platelet function of an EP3 recep-tor antagonist used alone and in combination with a P2Y12 antagonist both in-vitro and ex-vivo in human volunteers. Platelets 2013, 24, 392–400.
- Candish, L.; Mueller, S.; Suessmeier, F.; Lindner, N.; Gerdes, C.; Pook, E.; Buchmueller, A.; Gaugaz, F.Z.; Zimmermann, S.; Lang, D.; et al. Preparation of Substituted 4-oxo-3,4-dihydropyrrolotriazine-6-carboxamides as EP3 Receptor Antagonists. WO Patent WO2021094209A1, 20 May 2021.
- Zhang, M.; Jang, H.; Nussinov, R. PI3K inhibitors: Review and new strategies. Chem. Sci. 2020, 11, 5855–5865.
- Ellis, H.; Ma, C.X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110.
- Erra, M.; Taltavull, J.; Gréco, A.; Bernal, F.J.; Caturla, J.F.; Gràcia, J.; Domínguez, M.; Sabaté, M.; Paris, S.; Soria, S.; et al. Discovery of a Potent, Selective, and Orally Available PI3Kδ Inhibitor. ACS Med. Chem. Lett. 2017, 8, 118–123.
- Erra, M.; Taltavull, J.; Bernal, F.J.; Caturla, J.F.; Carrascal, M.; Pagès, L.; Mir, M.; Espinosa, S.; Gracia, J.; Dominguez, M.; et al. Discovery of a Novel Inhaled PI3Kδ Inhibitor for the Treatment of Respiratory Diseases. J. Med. Chem. 2018, 61, 9551–9567.
- Jia, H.; Dai, G.; Su, W.; Xiao, K.; Weng, J.; Zhang, Z.; Wang, Q.; Yuan, T.; Shi, F.; Zhang, Z.; et al. Discovery, Optimization, and Evaluation of Potent and Highly Selective PI3Kγ-PI3Kδ Dual Inhibitors. J. Med. Chem. 2019, 62, 4936–4948.
- Wang, Y.; Wu, X.; Du, M.; Chen, X.; Ning, X.; Chen, H.; Wang, S.; Liu, J.; Liu, Z.; Li, R.; et al. Eg5 inhibitor YL001 induces mitotic arrest and inhibits tumor proliferation. Oncotarget 2017, 8, 42510–42524.
- Kim, K.S.; Lu, S.; Cornelius, L.A.; Lombardo, L.J.; Borzilleri, R.; Schroeder, G.M.; Sheng, C.; Rovnyak, G.; Crews, D.; Schmidt, R.J.; et al. Synthesis and SAR of pyrrolotriazine-4-one based Eg5 inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 3937–3942.
- Fujita, M.; Scheffer, D.D.L.; Turnes, B.L.; Cronin, S.J.F.; Latrémolière, A.; Costigan, M.; Woolf, C.J.; Latini, A.; Andrews, N.A. Sepiapterin Reductase Inhibition Leading to Selective Reduction of Inflammatory Joint Pain in Mice and Increased Urinary Sepiapterin Levels in Humans and Mice. Arthritis Rheumatol. 2020, 72, 57–66.
- Tebbe, M.J.; Atton, H.V.; Avery, C.; Bromidge, S.M.; Kerry, M.; Kotey, A.K.; Monck, N.J.; Meniconi, M.; Ridgill, M.P.; Tye, H.; et al. Preparation of Heteroaryl Derivatives as Sepiapterin Reductase Inhibitors. WO Patent WO2017059191A1, 6 April 2017.
- Cartel, M.; Mouchel, P.L.; Gotanègre, M.; David, L.; Bertoli, S.; Mansat-De Mas, V.; Besson, A.; Sarry, J.E.; Manenti, S.; Didier, C. Inhibition of ubiquitin-specific protease 7 sensitizes acute myeloid leukemia to chemotherapy. Leukemia 2021, 35, 417–432.
- Ioannidis, S.; Talbot, A.C.; Follows, B.; Buckmelter, A.J.; Wang, M.; Campbell, A.-M. Preparation of Pyrrolotriazinones and Imidazotriazinones as Ubiquitin-Specific Protease 7 Inhibitors. U.S. Patent 20160185786A1, 19 June 2018.
More
Information
Subjects:
Chemistry, Medicinal
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
692
Revisions:
2 times
(View History)
Update Date:
17 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No