Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ana Luisa Chávez-Hernández | + 2659 word(s) | 2659 | 2021-12-06 09:14:53 | | | |
| 2 | Ana Luisa Chávez-Hernández | Meta information modification | 2659 | 2021-12-17 02:12:08 | | | | |
| 3 | Rita Xu | Meta information modification | 2659 | 2021-12-17 02:29:03 | | | | |
| 4 | Ana Luisa Chávez-Hernández | Meta information modification | 2659 | 2021-12-17 03:20:27 | | | | |
| 5 | Ana Luisa Chávez-Hernández | Meta information modification | 2659 | 2021-12-17 03:23:18 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Chávez-Hernández, A.L. De Novo Design of HIV-1 Protease Inhibitors. Encyclopedia. Available online: https://encyclopedia.pub/entry/17238 (accessed on 24 June 2026).
Chávez-Hernández AL. De Novo Design of HIV-1 Protease Inhibitors. Encyclopedia. Available at: https://encyclopedia.pub/entry/17238. Accessed June 24, 2026.
Chávez-Hernández, Ana Luisa. "De Novo Design of HIV-1 Protease Inhibitors" Encyclopedia, https://encyclopedia.pub/entry/17238 (accessed June 24, 2026).
Chávez-Hernández, A.L. (2021, December 17). De Novo Design of HIV-1 Protease Inhibitors. In Encyclopedia. https://encyclopedia.pub/entry/17238
Chávez-Hernández, Ana Luisa. "De Novo Design of HIV-1 Protease Inhibitors." Encyclopedia. Web. 17 December, 2021.
Copy Citation
Acquired immunodeficiency syndrome (AIDS) caused by the human immunodeficiency virus (HIV) continues to be a public health problem. In 2020, 680,000 people died from HIV-related causes, and 1.5 million people were infected. Antiretrovirals are a way to control HIV infection but not to cure AIDS.
artificial intelligence
de novo design
fragment-based drug discovery
HIV-1 inhibitors
1. Introduction
The acquired immunodeficiency syndrome (AIDS) caused by the human immunodeficiency virus (HIV) is a major global public health concern. In 2020, the World Health Organization (WHO) reported that approximately 37.7 million people live with HIV out of 24.5 million from the African region. In 2020, 680,000 people died from HIV-related causes and 1.5 million people acquired it[1]. There is no definite treatment for AIDS. Therefore, it is necessary to collaborate to develop a treatment since the antiretroviral drugs currently approved by Food and Drug Administration (FDA) to clinical use only control AIDS and prevent HIV-1 transmission between individuals (Figure 1 and Table 1)[2][3][4].
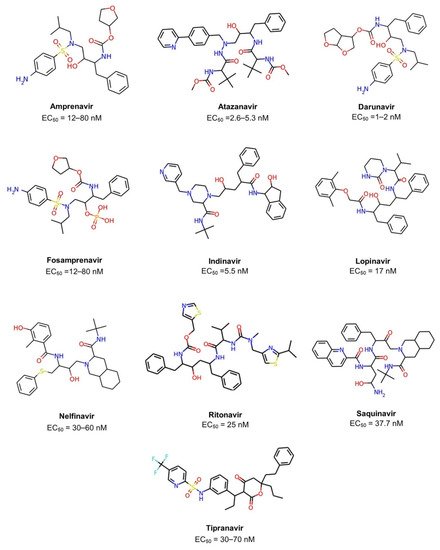
Figure 1. Chemical structures of ten FDA-approved HIV-1 protease inhibitors (Amprenavir, Atazanavir, Darunavir, Fosamprenavir, Indinavir, Lopinavir, Nelfinavir, Ritonavir, Saquinavir, Tipranavir). The EC50 is the concentration of drug required to produce 50% of the maximum possible effect.
Table 1. FDA-approved HIV-1 protease inhibitors which will be used as a reference for the de novo design of the new chemical compounds. a Fosamprenavir is the phosphate ester prodrug of amprenavir.
| Generic Name | Brand Name | EC50[3] | FDA Approval |
|---|---|---|---|
| Amprenavir | Agenerase | 12–80 nM | 1999 |
| Atazanavir | Reyataz | 2.6–5.3 nM | 2003 |
| Darunavir | Prezista | 1–2 nM | 2006 |
| Fosamprenavir a | Lexiva | 12–80 nM | 2003 |
| Indinavir | Crixivan | 5.5 nM | 1996 |
| Lopinavir | Kaletra | 17 nM | 2000 |
| Nelfinavir | Viracept | 30–60 nM | 1997 |
| Ritonavir | Norvir | 25 nM | 1996 |
| Saquinavir | Invirase | 37.7 nM | 1995 |
| Tipranavir | Aptivus | 30–70 nM | 2005 |
Drug design and development demand many years of hard work and economic investment. Most drug candidates are prone to fail [5]. From 25,000 compounds that start in the laboratory, only 25 make it through preclinical testing to human testing, and just five of those reach the actual clinical use[6]. Computer-aided drug design (CADD) has contributed to yielding several drugs into the clinic, yet it has several challenges ahead [7]. Among the CADD methods, de novo design has gained relevance due to the diversity of structures generated by optimizing the algorithms used. From a methodological point of view, artificial intelligence as boosted the development and application of de novo design[5][8][9]. Notably, de novo design is a structure-based drug design method that benefits from the experimental information available of the binding sites of molecular targets.
The main goal of de novo design is to suggest novel molecular structures from scratch with desired activity on a pharmacological target and desired properties[10]. The new structures can be made using two general approaches: fragment-based and atom-based. The advantage of the fragment-based approach is that it narrows down the search in chemical space and maintains good chemical structure diversity[11][12][13]. Additionally, fragments form fewer interactions that should be able to bind to a greater number of sites on a greater number of proteins. Fragments are small (less than 20 heavy atoms) and typically soluble; they are likely to have better pharmaceutical properties as well as the new chemical compounds generated from them[14]. Over the last 20 years, four drugs from fragment-based drug discovery (FBDD) have been approved, and 40 compounds are currently in clinical trials [15].
Recently, de novo design and artificial intelligence have been combined to propose novel molecules for the treatment of SARS-CoV-2 based on HIV-1 protease and the approved drugs that inhibit this viral protease[8]. Another successful example of de novo design focusing on HIV research led to four molecules from a new compound library generated from the ZINC database[16]. Other approaches de novo design was based on enumerating libraries using chemical reactions[17][18] and are also promising to expand the epigenetic relevant chemical space[19].
The development of new chemical compounds using de novo design can begin from natural product-derived fragments. Natural products have been attractive chemical compounds because they are characterized by a larger number of sp3 carbon atoms, chiral centers (associated with structural complexity), the larger scaffold diversity, and functional groups, hence their relevance for use as building-blocks[20][21]. Indeed, larger structural complexity of small organic molecules has been associated with increased selectivity and drug-likeness. In previous studies, we showed that natural products cover regions of chemical space that have not yet been explored by synthetically accessible compounds and those with biological activity[22]. For this reason, natural products could be used as building-blocks to develop novel synthetic molecules or pseudo-natural products which combine the desired structural characteristics from different natural products[23].
2. Chemical Space Visualization
A visual representation of the chemical space based on physicochemical properties (MW, HB, HBA, SlogP, TPSA, and RB) using PCA is shown in Figure 2. Principal component 1 recovered 73.6% of the variance, and principal component 2 recovered 21.2% of the variance. The accumulated variance recovered by the first two principal components represented in Figure 2 was 94.8%. In this chemical space visualization, the compounds generated from the three fragment libraries are within the space of physicochemical properties of FDA-approved drugs. Likewise, some compounds generated from COCONUT fragments had physicochemical properties similar to FDA-approved HIV-1 protease.
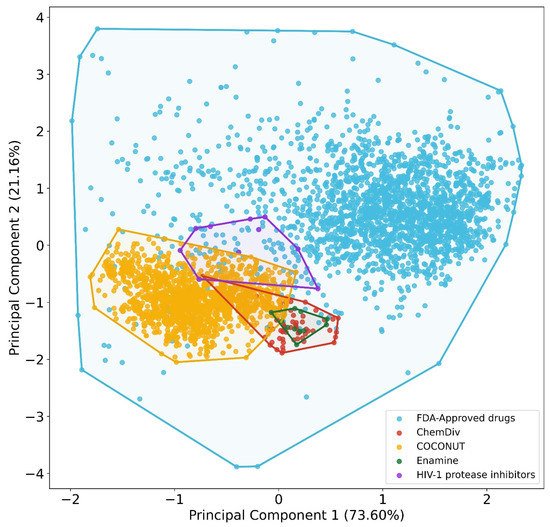
Figure 2. Chemical space visualization of the virtual focused compound library of HIV-1 viral protease inhibitors from natural product fragments and two compound reference libraries using PCA based on physicochemical properties. Compound reference libraries represented in colors: FDA-approved drugs (blue) and FDA-approved HIV-1 protease inhibitors (purple). Likewise. for new chemical compounds generated from COCONUT (orange), ChemDiv (red), and Enamine (green) fragment libraries.
To quantitatively define which dataset is the most diverse, coverage space obtained by convex hull analysis derived from PCA was computed for each dataset. The convex hull is defined as the minimum convex polygon so that the point set is either inside this polygon or at its border[24][25]. The convex hull area computed were for FDA-approved drugs (737.59), HIV-1 protease inhibitors (1.11), compounds from COCONUT’s fragments (3.18), compounds from ChemDiv’s fragments (0.79), and compounds from Enamine fragments (0.18). The outcome of this analysis was similar to the results of the structural diversity analysis based on fingerprints: reference datasets were more diverse than the new chemical compounds generated from fragments datasets. The new chemical compounds derived from COCONUT fragments were the most diverse, followed by new chemical compounds derived from ChemDiv and Enamine fragments.
The visual representation of the chemical space based on molecular fingerprint using the TMAP algorithm is shown in Figure 3. An interactive version of the TMAP is available at https://figshare.com/s/ceb58d58e8f5585ce67e (accessed on 5 November 2021). The chemical structures of new chemical compounds generated were very different in comparison with FDA-approved drugs and FDA-HIV-1 protease inhibitors. The chemical structures of the new compounds generated from ChemDiv and Enamine fragments were very similar compared to compounds derived from COCONUT fragments. In some cases, the chemical structures of compounds generated from COCONUT’s fragments were very similar to some FDA-approved drugs, for instance, palbociclib and pipecuronium. In these cases where there are not commercially available fragments like COCONUT’s fragments could be used palbociclib and pipecuronium.
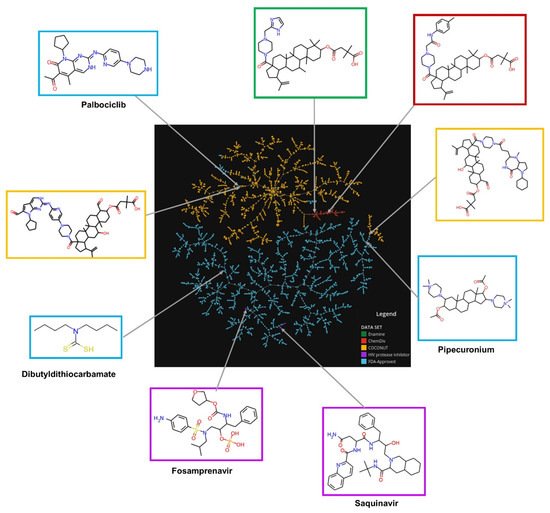
Figure 3. Chemical space visualization of the virtual focused compound library of HIV-1 viral protease inhibitors from natural product fragments and two compound reference libraries using TMAP based on molecular fingerprints. Compounds reference libraries represented in colors: FDA-approved drugs (blue), and FDA-approved HIV-1 protease inhibitors (purple). Likewise, for new chemical compounds generated from COCONUT (orange), ChemDiv (red), and Enamine (green) fragment libraries. The interactive version is available at https://figshare.com/s/ceb58d58e8f5585ce67e (accessed on 5 November 2021).
3. Compound Filtering Based on Physicochemical Properties
Figure 4 shows box-whisker plots of physicochemical properties after applying the empirical rules proposed. 352 compounds generated from COCONUT fragments (20%) and 1 compound generated from ChemDiv fragments were retained (2%), and compounds generated from Enamine fragments were not retained (0%). Based on the properties’ distribution shown in the box-whisker plots, the physicochemical properties of compounds generated from COCONUT fragments, ChemDiv fragments, and Enamine fragments were different regarding FDA-approved HIV-1 protease inhibitors and FDA-approved drugs.
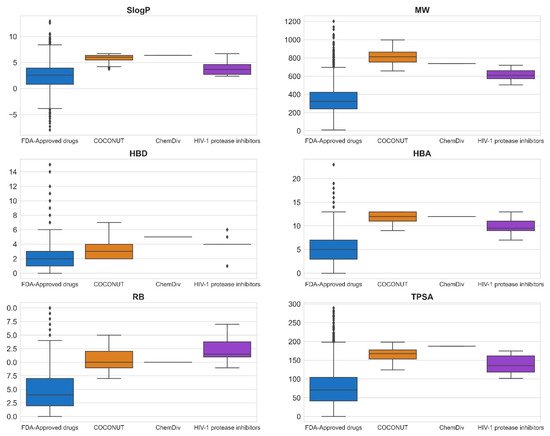
Figure 4. Box-whisker plots of physicochemical properties of FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple), and new chemical compounds generated from COCONUT (orange) and ChemDiv (red) fragment libraries after applying physicochemical properties filtering. Black diamonds represent outliers.
The physicochemical properties calculated for datasets were: SlogP ≤ 12.94, MW ≤ 1201.84, RB ≤ 20, TPSA ≤ 286.50, HBA ≤ 23, HBD ≤ 15 for FDA-approved drugs; SlogP ≤ 6.70, MW ≤ 720.31, RB ≤ 17, TPSA ≤ 174.56, HBA ≤ 13, HBD ≤ 6 for FDA-approved HIV-1 protease inhibitors; SlogP ≤ 6.69, MW ≤ 998.63, RB ≤ 15, TPSA ≤ 198.54, HBA ≤ 13, HBD ≤ 7 for compounds generated from COCONUT fragments, and SlogP = 6.4, MW = 737.47, RB = 10, TPSA = 187.47, HBA = 12, HBD = 5 for the compound generated from ChemDiv’s fragments. The SlogP, RB, and HBA values of compounds generated from COCONUT fragments and ChemDiv fragments were less than FDA-approved HIV-1 protease inhibitors. HBA values were equal or less than FDA-approved HIV-1 protease inhibitors. The SlogP values of compounds derived from Enamine fragments were larger than FDA-approved HIV-1 protease inhibitors; accordingly, no compound was retained. The MW, TPSA, and HBD values of compounds generated from COCONUT fragments were larger than for FDA-approved HIV-1 protease inhibitors and less than for FDA-approved drugs. As mentioned above Ganesan[26], natural products that violate the Lipinsky rules remain largely compliant in terms of log P and HBD. He considers that “nature has learned to maintain low hydrophobicity and intermolecular H-bond donating potential when it needs to make biologically active compounds with high molecular weight and a large number of rotatable bonds”. In drugs, the molecules that exceed HBD 5 or HBA 10 the majority are natural product-related[27].
4. Filtering Based on Synthetic Feasibility
The synthetic feasibility was computed for FDA-approved drugs, FDA-approved HIV-1 protease inhibitors, and compounds generated from COCONUT and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors. Figure 5 summarizes the results of synthetic feasibility. Molecules with a low SAscore value < 6 are easily synthetically accessible [28]. A total of 97% FDA-approved drugs had SAscore < 6, and FDA-approved HIV-1 protease inhibitors had SAscore ≤ 4.24. Similarly, 75% of compounds generated from COCONUT fragments had SAscore ≤ 6.03 and the compound generated from ChemDiv had SAscore = 5.54. Although, compounds generated from COCONUT fragments had 5.50 ≤ SAscore ≤ 6.03, still in recommended range so that can be synthetically accessible; moreover, the high SAscore, in compounds generated regarding FDA-approved HIV-1 protease inhibitors, was influenced by the ten stereocenters of betulinic acid and 24-nor-3α,11α-dihydroxy-lup-20(29)-en-23,28-dioic acid. Considering that these stereocenters do not have to be generated within the organic synthesis, the SAscore value would be lower.
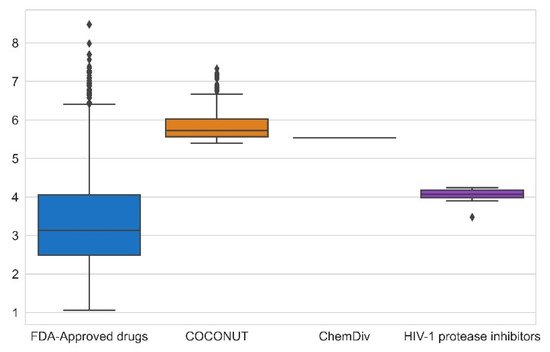
Figure 5. Box-whisker plot of synthetic feasibility calculated for FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple), and new chemical compounds generated from COCONUT fragments (orange) and ChemDiv (red) fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors. Black diamonds represent outliers.
5. ADME-Tox Profiling
The ADME-Tox profiling was computed for 251 compounds generated from COCONUT fragments and 1 compound generated from ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and estimated as easy synthesizable (i.e., SAscore ≤ 6). Similarly, ADME-Tox profiling was computed for FDA-approved drugs and FDA-approved HIV-1 protease inhibitors.
5.1. Absorption
Solubility, lipophilicity, and HIA are summarized in Figure 6. Solubility was expressed by Silicos-IT LogSw and lipophilicity was expressed by consensus LogP. Silicos-IT LogSw and consensus LogP were computed with the SwissADME server. Percentage of HIA was computed with the pkCSM-pharmacokinetics server.
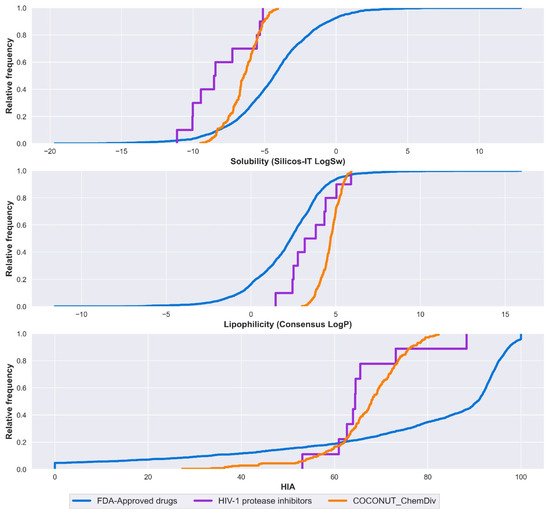
Figure 6. Distribution curve of solubility, lipophilicity, and HIA. Colors represent compounds: new chemical compounds generated from COCONUT fragments and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and easily synthetically accessible (orange), FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple). Solubility is expressed in the percentage of Silicos-IT LogSw, and lipophilicity is expressed in the percentage of consensus LogP.
Median values for solubility, lipophilicity, and HIA are described below. FDA-approved drugs had consensus LogP = 2.36, Silicos-IT LogSw = −4.34, HIA = 90.6%. FDA-approved HIV-1 protease inhibitors had consensus LogP = 3.50, Silicos-IT LogSw = −8.49, HIA = 64.4%. Compounds derived from COCONUT and ChemDiv had consensus LogP = 4.70 and Silicos-IT LogSw = −6.45, HIA = 67.9%.
New drug candidates have poor water solubility, and it is often the result of highly lipophilic compounds. Log P < 2, the crystal lattice becomes the main determining factor for solubility. LogP values above 2, the lipophilicity is the main factor [29]. FDA-approved HIV-1 protease inhibitors were highly soluble, followed by compounds derived from COCONUT and ChemDiv fragments, both had Log P > 2; in this case, solubility is strongly influenced by lipophilicity. Contrary to FDA-approved drugs that had Log P close to 2 and were less soluble, solubility mainly depends on the crystal lattice. Compounds derived from COCONUT and ChemDiv fragments had higher HIA in comparison to FDA-approved HIV-1 protease inhibitors.
5.2. Distribution
The relative frequency of BBB permeability is described in Figure 7. The median value of BBB permeability was −0.38 for FDA-approved drugs; −1.21 for compounds generated from COCONUT and ChemDiv fragments, and −1.25 for FDA-approved HIV-1 protease inhibitors. Compounds generated from COCONUT and ChemDiv fragments had similar BBB permeability.
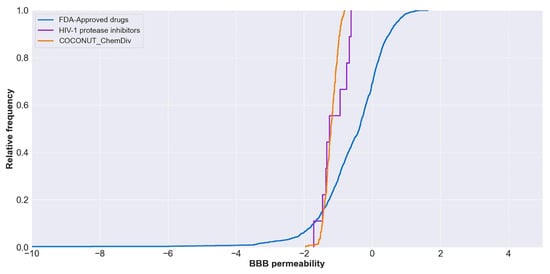
Figure 7. Distribution curve of BBB permeability. Colors represent compounds: new chemical compounds generated from COCONUT fragments and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and easily synthetically accessible (orange), FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple). The BBB permeability of FDA-approved drugs was between −34 and 2.
The percentage of compounds that are P-glycoprotein substrate, P-glycoprotein I inhibitor, and P-glycoprotein II inhibitor were summarized in Figure 8. All FDA-approved HIV-1 protease inhibitors and 96% of compounds generated from COCONUT and ChemDiv fragments were P-glycoprotein substrates. Similarly, 66.67% of HIV-1 Approved protease inhibitors and 82.9% of compounds generated from COCONUT and ChemDiv fragments were P-glycoprotein II inhibitors. Whereas no compounds generated from COCONUT and ChemDiv fragments were P-glycoprotein I inhibitors, against 100% FDA-approved HIV-1 proteases inhibitors were P-glycoprotein I inhibitors.
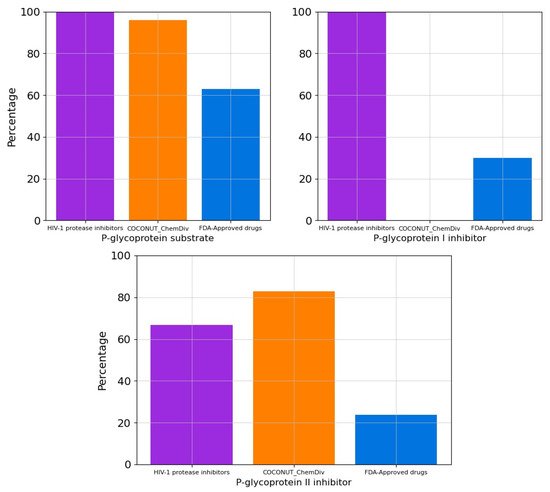
Figure 8. Percentage of compounds that are P-glycoprotein substrate, P-glycoprotein I inhibitor, and P-glycoprotein II inhibitor. Colors represent compounds: new chemical compounds generated from COCONUT fragments and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and easily synthetically accessible (orange), FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple).
5.3. Metabolism
The percentage of compounds CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 inhibitors is described in Figure 9. No compounds generated from COCONUT and ChemDiv fragments were CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4 inhibitors. FDA-approved HIV-1 inhibitors were not CYP1A2 and CYP2D6 inhibitors similar to compounds generated from COCONUT and ChemDiv fragments. Whereas for FDA-approved HIV-1protease inhibitors, 89% were CYP3A4 inhibitors, and 33% were CYP2C19 and CYP2C9 inhibitors.
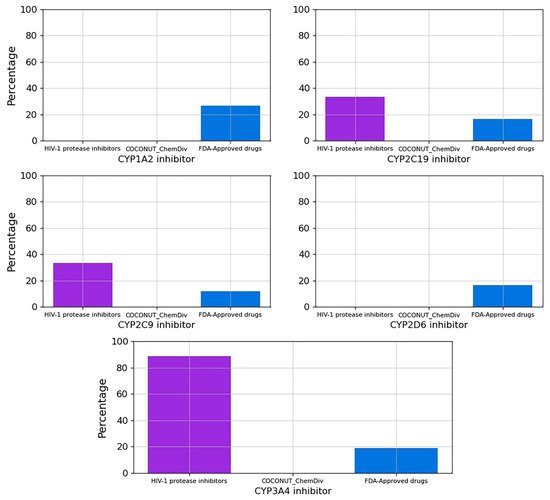
Figure 9. Percentage of compounds that inhibit the main cytochromes, CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4. Colors represent compounds: new chemical compounds generated from COCONUT fragments and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and easily synthetically accessible, FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple).
5.4. Excretion
Clearance quantitates the irreversible removal of a drug from the measured matrix, generally, blood or plasma[30]. The total clearance logarithm expressed in units of (mL/min/Kg) is shown in Figure 10. The median values of the total clearance logarithm were 0.591 for FDA-approved drugs; 0.494 for FDA-approved HIV-1 protease inhibitors, and −0.618 for compounds derived from COCONUT and ChemDiv fragments. The total clearance of FDA-approved HIV-1 protease inhibitors (0.20 ≤ total clearance ≤ 0.94) was similar to 75% FDA-approved drugs (0.27 ≤ total clearance ≤ 0.85). Whereas the total clearance of compounds generated from COCONUT and ChemDiv fragments (−1.34 ≤ total clearance ≤ 0.13) was similar to 25% FDA-approved drugs (-13.94 ≤ total clearance ≤ 0.27). The total clearance of compounds derived from COCONUT and ChemDiv fragments and FDA-approved HIV-1 inhibitors were different.
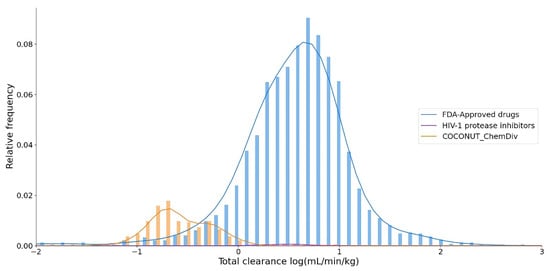
Figure 10. Distribution curve of the total clearance. Colors represent compounds: new chemical compounds generated from COCONUT fragments and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and easily synthetically accessible (orange), FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple).
5.5. Toxicity
Percentage of compounds from datasets that are hERG I inhibitor, hERG II inhibitor, hepatotoxicants (hepatotoxicity), and carcinogens (positive in AMES test) were described in Figure 11. FDA-approved HIV-1 protease inhibitors and compounds generated from COCONUT and ChemDiv fragments were not carcinogens. However, 77.22% of compounds derived from COCONUT and ChemDiv fragments were hepatotoxicants, lower than FDA-approved HIV-1 protease inhibitors (100%), and higher than FDA-approved drugs (47.42%). A total of 100% and 98.81% of compounds generated from COCONUT and ChemDiv fragments were not hERG I/II inhibitors, respectively.
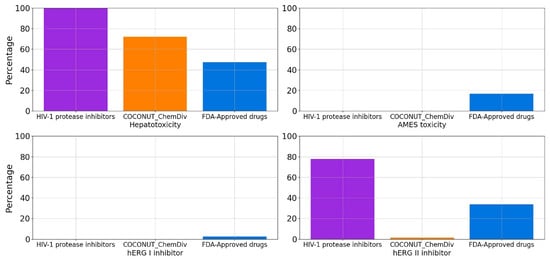
Figure 11. Percentage of compounds that are hERG I inhibitor, hERG II inhibitor, hepatotoxicity, and toxicity in AMES test in silico. Colors represent compounds: new chemical compounds generated from COCONUT fragments and ChemDiv fragments with physicochemical properties like FDA-approved HIV-1 protease inhibitors and easily synthetically accessible (orange), FDA-approved drugs (blue), FDA-approved HIV-1 protease inhibitors (purple).
References
- HIV/AIDS. Available online: https://www.who.int/news-room/fact-sheets/detail/hiv-aids (accessed on 15 July 2021).
- Zulfiqar, H.F.; Javed, A.; Sumbal; Afroze, B.; Ali, Q.; Akbar, K.; Nadeem, T.; Rana, M.A.; Nazar, Z.A.; Nasir, I.A.; et al. HIV Diagnosis and Treatment through Advanced Technologies. Front. Public Health 2017, 5, 32.
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: A review of molecular selectivity and toxicity. HIV AIDS 2015, 7, 95–104.
- FDA. Available online: https://www.fda.gov/consumers/free-publications-women/hiv-and-aids-medicines-help-you (accessed on 28 April 2021).
- Schneider, G.; Clark, D.E. Automated de novo drug design: Are we nearly there yet? Angew. Chem. Int. Ed. 2019, 58, 10792–10803.
- Torjesen, I. Drug Development: The Journey of a Medicine from Lab to Shelf. Available online: https://pharmaceutical-journal.com/article/feature/drug-development-the-journey-of-a-medicine-from-lab-to-shelf (accessed on 29 May 2021).
- Medina-Franco, J.L. Grand challenges of computer-aided drug design: The road ahead. Front. Drug Discov. 2021, 1, 728551.
- Bung, N.; Krishnan, S.R.; Bulusu, G.; Roy, A. De novo design of new chemical entities for SARS-CoV-2 using artificial intelligence. Future Med. Chem. 2021, 13, 575–585.
- Liu, X.; IJzerman, A.P.; van Westen, G.J.P. Computational approaches for de novo drug design: Past, present, and future. In Artificial Neural Networks; Cartwright, H., Ed.; Springer: New York, NY, USA, 2021; pp. 139–165. ISBN 978-1-0716-0826-5.
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676.
- Meyers, J.; Fabian, B.; Brown, N. De novo molecular design and generative models. Drug Discov. Today 2021, 26, 2707–2715.
- Devi, R.V.; Sathya, S.S.; Coumar, M.S. Evolutionary algorithms for de novo drug design—A survey. Appl. Soft Comput. 2015, 27, 543–552.
- Hartenfeller, M.; Schneider, G. De Novo Drug Design. In Chemoinformatics and Computational Chemical Biology; Bajorath, J., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 299–323. ISBN 978-1-60761-839-3.
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug Discov. 2016, 15, 605–619.
- Osborne, J.; Panova, S.; Rapti, M.; Urushima, T.; Jhoti, H. Fragments: Where are we now? Biochem. Soc. Trans. 2020, 48, 271–280.
- Shinde, P.B.; Bhowmick, S.; Alfantoukh, E.; Patil, P.C.; Wabaidur, S.M.; Chikhale, R.V.; Islam, M.A. De novo design based identification of potential HIV-1 integrase inhibitors: A pharmacoinformatics study. Comput. Biol. Chem. 2020, 88, 107319.
- Ghiandoni, G.M.; Bodkin, M.J.; Chen, B.; Hristozov, D.; Wallace, J.E.A.; Webster, J.; Gillet, V.J. Enhancing reaction-based de novo design using a multi-label reaction class recommender. J. Comput. Aided Mol. Des. 2020, 34, 783–803.
- Saldívar-González, F.I.; Huerta-García, C.S.; Medina-Franco, J.L. Chemoinformatics-based enumeration of chemical libraries: A tutorial. J. Cheminform. 2020, 12, 64.
- Prado-Romero, D.L.; Medina-Franco, J.L. Advances in the exploration of the epigenetic televant chemical space. ACS Omega 2021, 6, 22478–22486.
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; International Natural Product Sciences Taskforce; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216.
- Barnes, E.C.; Kumar, R.; Davis, R.A. The use of isolated natural products as scaffolds for the generation of chemically diverse screening libraries for drug discovery. Nat. Prod. Rep. 2016, 33, 372–381.
- Chávez-Hernández, A.L.; Sánchez-Cruz, N.; Medina-Franco, J.L. A Fragment library of natural products and its comparative chemoinformatic characterization. Mol. Inform. 2020, 39, 2000050.
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235.
- Saldívar-González, F.I.; Lenci, E.; Calugi, L.; Medina-Franco, J.L.; Trabocchi, A. Computational-aided design of a library of lactams through a diversity-oriented synthesis strategy. Bioorg. Med. Chem. 2020, 28, 115539.
- Laurini, R. 5—Geographic Relations; Laurini, R.B.T.-G.K.I., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 83–109. ISBN 978-1-78548-243-4.
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317.
- Tinworth, C.P.; Young, R.J. Facts, Patterns, and principles in drug discovery: Appraising the Rule of 5 with measured physicochemical data. J. Med. Chem. 2020, 63, 10091–10108.
- Ertl, P.; Schuffenhauer, A. Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J. Cheminform. 2009, 1, 8.
- Bergström, C.A.S.; Yazdanian, M. Lipophilicity in drug development: Too much or not enough? AAPS J. 2016, 18, 1095–1100.
- Smith, D.A.; Beaumont, K.; Maurer, T.S.; Di, L. Clearance in drug design. J. Med. Chem. 2019, 62, 2245–2255.
More
Information
Subjects:
Medical Informatics
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.0K
Revisions:
5 times
(View History)
Update Date:
17 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No