Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carmelo Gurnari | + 1924 word(s) | 1924 | 2021-12-14 03:56:24 | | | |
| 2 | Jessie Wu | Meta information modification | 1924 | 2021-12-16 02:22:50 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Gurnari, C. Alternative Splicing in Myeloid Malignancies. Encyclopedia. Available online: https://encyclopedia.pub/entry/17170 (accessed on 09 August 2026).
Gurnari C. Alternative Splicing in Myeloid Malignancies. Encyclopedia. Available at: https://encyclopedia.pub/entry/17170. Accessed August 09, 2026.
Gurnari, Carmelo. "Alternative Splicing in Myeloid Malignancies" Encyclopedia, https://encyclopedia.pub/entry/17170 (accessed August 09, 2026).
Gurnari, C. (2021, December 15). Alternative Splicing in Myeloid Malignancies. In Encyclopedia. https://encyclopedia.pub/entry/17170
Gurnari, Carmelo. "Alternative Splicing in Myeloid Malignancies." Encyclopedia. Web. 15 December, 2021.
Copy Citation
Alternative RNA splicing (AS) is an essential physiologic function that diversifies the human proteome. AS also has a crucial role during cellular development. In fact, perturbations in RNA-splicing have been implicated in the development of several cancers, including myeloid malignancies.
RNA-splicing
cancer
therapies
1. Introduction
In the last 10–15 years, advances in genomic technologies and deep RNA-sequencing in combination with whole-genome sequencing have enormously increased the chance to identify a global dysregulation in RNA-splicing and its association with gene mutations in hematologic malignancies [1]. Pre-mRNA splicing is a precise process in which non-coding sequences of a gene are excised and coding regions are joined together to form a mature RNA for subsequent protein translation [2]. Alternative splicing (AS) is a natural process creating transcript variability, and it is common to 95% of human genes. Splicing diversity can either be physiologic and present in normal individuals without leading to pathologic conditions or be potentially selective to induce human diseases, often resulting in the production of cancer vulnerability due to abnormal AS phenomena [3]. Indeed, aberrant AS produced at a single cell level can impair the subsequent cell development if occurring in key transcriptional factors crucial to assign cell identity [4][5]. Moreover, a variety of chemotherapeutic drugs can induce changes in AS modalities, typically intron retention, which is similar among different pharmacologic agents and produces a down-regulation of gene expression and phosphorylation [6]. Several modes of AS are represented in Figure 1.
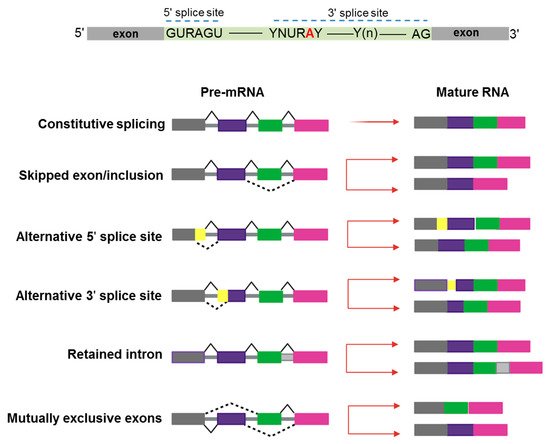
Figure 1. Schematic representation of constitutive and alternative splicing. Upper panel: 5′ and 3′ splice-site and branch-site consensus sequences are shown for mammalian U2-dependent introns. R indicates a purine, Y a pyrimidine, and N any other nucleotide. The branch point adenosine is indicated in red color.A tract of 10 to 20-nucleotides forms a pyrimidine tract (Yn) between the branch site and 3′ splice site. Modes of action of RNA-splicing from pre-messenger RNA to mature RNA are presented. Canonical transcripts are produced via constitutive splicing, while variability is naturally produced through different modes of alternative splicing.
2. Alternative Splicing as a Novel Mechanism of Neo-Antigens Production
Perturbations in RNA-splicing due to mutations in SF genes constitute also a modality that generates tumor neo-antigens. These neo-antigens can be private (specific to single patients), public (shared across patients) and disease-specific (not present in normal tissues of normal counterparts). Cancer-specific AS has been studied across large datasets of cancer types with the purpose of identifying shared novel exon-junctions. However, many novel exon-junctions are still biologically and functionally unexplored [7]. In-depth integrated analyses of RNA and whole-exome sequencing of tumors from 8705 donors, including 670 matched normal samples (TCGA and GTEx databases) from a variety of cancer types have shown that mutations in certain SFs (for instance, SF3B1, U2AF1) might generate immunogenic neo-antigens [8][9]. In several cases, mutations in SF genes led to abnormal open reading frames, retention of intronic sequences and in-frame insertions of codons, possibly forming neo-epitopes. Indeed, neo-junction-derived peptides were identified as putative neo-antigens in disease samples and verified based on bioinformatics prediction tools and mass-spectrometry. These studies, together with others [10][11], pointed out not only the frequency and abundance of novel splicing events but also the lack of recurrence and restriction of neo-antigens due to the high diversity in HLA haplotypes. Furthermore, neo-antigens may also be the product of errors in mis-splicing of exons and transcription of microsatellites, specifically indels, which, in normal conditions, are eliminated by NMD and not presented to the immune system while in cancer cells are found on the surface of antigen-presenting cells (APC) linked to MCH class I and II molecules [12].
RNA-sequencing studies conducted in myeloid malignancies (MDS and AML) have shown that AS can generate novel protein junctions also through gain and loss of coding RNA sequences or by the production of frameshifts. These novel coding regions, when identified in patient samples, might potentially help the final diagnosis of some myeloid disorders, such as MDS, whose diagnostic criteria are sometimes relatively subjective (e.g., dysplasia assessment) [13]. In this study, specific disease-splicing events were classified as being selected for patients and not present in control samples. By testing for binding to class I MHC, 925 disease-specific splicing events were found. About 2% of these could be related to patients with one or more SF mutation, and some were consistently found in SF3B1 (FAM143A, FMNL1, PILRB) and SRSF2 (CD2BP2, RASGRP2) mutant cases. Studies conducted in induced pluripotent stem cells generated from MDS patient cells showed that SF3B1 mutations can produce HLA-presented neo-antigens, opening a new avenue for targeted immunotherapy. Interestingly, using HLA-binding algorithms (netMHCpan 4.0), peptides derived from mutated transcripts of SF3B1 showed a strong affinity for some class I HLA molecules (i.e., HLA-B*40:01), and their processing, presentation, and immunogenicity were functionally proven in vitro [14]. In particular, it has been demonstrated that certain cancers tend to present more neojunctions than others, irrespective of the observed mutational burden [9]. As discussed before, this is also true for MDS and AML, which present a high percentage of neosplicing events irrespective of SF mutations [13]. Besides mutation-produced neo-antigens, noncoding transcripts have also an immunogenic potential. Indeed, a combination of liquid chromatography tandem-mass spectrometry and RNA-sequencing has been used to detect noncoding transcripts, which can be created by introns, untranslated regions, and noncoding exons [15]. Splice-site-creating mutations are, in fact, predicted to generate an average of 2–2.5 neo-antigens per mutation [16]. In recent years, several workflows have been tested for the classification of these neo-antigens [17], and the optimization of bioinformatic tools has been growing extensively. By using RNA-sequencing for personalized neo-antigen prediction, a seminal study from Schischlik et al. [18] explored the neo-antigen repertoire in myeloid malignancies to find clues as to new targets for immunotherapy-based treatments. The neo-antigens repertoire was analyzed according to the presence and types of mutations, indels, fusion genes, and RNA-splicing errors. The majority of the mutations were of frameshift origin and caused the production of novel sequences, which were then analyzed for their binding affinity to MHC classes I and II. It is, thus, understandable how consequences of aberrant splicing, together with neo-antigens derived from mutated oncogenic proteins, in cancer cells may have a direct impact on the shape of the immune-peptidome presented to T cell effectors, finally contributing to mechanisms of immunoediting and clonal selection [19]. A summary of the role of mechanisms of AS in physiologic and pathologic conditions is presented in Figure 2.
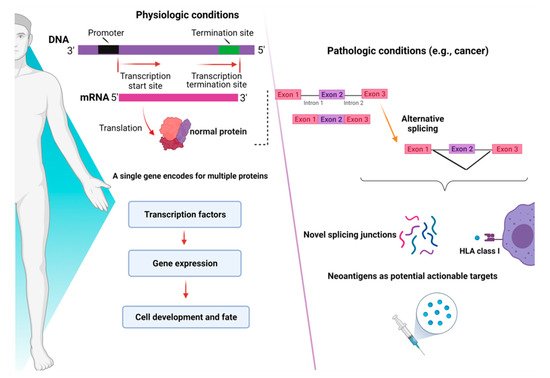
Figure 2. Consequences of alternative splicing in physiologic and pathologic conditions. In physiologic conditions (left panel), alternative splicing serves as a modulator of gene expression and regulates the function of transcriptional factors, ultimately dictating cell development and fate. In pathologic conditions (right panel), alternative splicing can produce novel junctions that, when exclusive to tumor cells, might represent actionable targets for immunotherapy. Images were generated (in part) using BioRender.
3. Antisense Oligonucleotides (ASOs)
ASOs are oligonucleotides of 15–25 bases in length and are reverse complement sequences of a specific RNA transcript region [20]. ASOs bind target RNA and preclude the direct access of SFs to the RNA target sequence or block/ silence enhancer elements of SFs, ultimately causing SFs to use alternative and proximal sequences. Several ASOs have been synthetically constructed to create loops to specifically block SFs or to target only mutant alleles in order to enhance selectivity and prevent alteration of normal sequences. Some studies have shown increased stability and simple internalization [1]. More recently, ASOs have also been shown to block the abnormal inclusion of a “poison exon” of BRD9 in SF3B1 mutant cells [21].
Efforts in drug development have been made to generate ASOs inducing RNase H1-mediated cleavage of target RNAs. This is the case of the gapmer ASOs composed of an oligodeoxynucleotide portion flanked by 2′ modified nucleotides at both ends. RNase H1-dependent ASOs are able to reduce levels of both cytoplasmic mRNA and retained nuclear RNA. The differences in ASOs activity in both nuclear and cytoplasmic compartments are due to many factors, including not only the presence of RNAseH1 but also specific RNA sequences and structure, RNA stability, and levels of ASOs (Figure 3) [22].
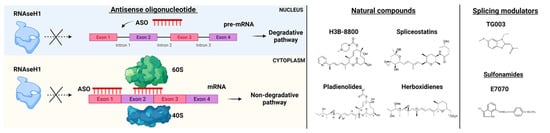
Figure 3. RNA-splicing-based targeted therapies. On the left, Antisense oligonucleotide (ASO) mechanisms of action. ASOs interfere with pre-mRNA by preventing RNaseH1 functions and blocking the interaction of mRNA with ribosomes (subunits 60S and 40S). On the right, examples of natural compounds, splicing modulators and sulfonamides used in pre-clinical and clinical models. Parts of the images were generated with BioRender.
Next-generation ASOs have hybrid structure antisense base-pairing sequences, which are linked to canonical binding sites of an SF. Further, ASOs have been constructed to induce usage of a cryptic ss or exon skipping. Novel methodologies incorporate ASOs in trans-splicing technology or Spliceosomal-Mediated RNA trans-splicing (SMaRT), which replaces the entire coding sequence of a target ss through ASO-containing plasmids targeting an endogenous intron of the mutant ss. This method has been used to correct mutations occurring in the 5′ ss or 3′ ss [20]. For instance, this approach has been applied to replace the first exon of the HBB (β-globin) gene in β-thalassemia [23]. Splicing restoration has also been tested using different versions of the spliceosomal snRNAs designed to base-pairing in a mutant 5′ ss. Furthermore, the introduction of genome editing technologies, including transcription activator-like nucleases (TALENs) and CRISPR/Cas9, have been implemented to correct splicing defects [24]. In fact, CRISPR/Cas9 was used to edit protein domains and identify drug targets by screening 192 chromatin regulatory domains in MLL-AF9/NrasG12D models of AML [25]. Other groups applied a CRISPR/Cas9 method to edit multiple genes mutated in AML and track clonal dynamics in murine systems using Lin–Sca1+c-Kit+ (LSK) cells [26].
4. Small Molecules Agents
Small molecules have been tested in several models of spliceosome dysfunction. Specifically, H3B-8800, a selective and orally bioavailable modulator of normal and mutant SF3b complex, has shown dose-dependent modulation of splicing in pre-clinical xenograft models [27], but unfortunately, these data have not been confirmed by the phase I, first-in-human open-label, multicenter trial for patients with previously treated MDS, AML, and CMML (NCT02841540) [28]. The discovery of H3B-8800 follows a path of research efforts on natural products (Figure 3). Indeed, products derived from bacteria have been shown to bind the SF3b complex and disrupt the early assembly of the spliceosome. These compounds have different pharmacologic activities and include low (FR901463, FR901464, FR901465, herboxidienes, and pladienolides) and high stability agents (E7107, a pladienolide B-analog, spliceostatin A, and the FR901464 derivative sudemycins). While several agents have only been shown to biologically alter splicing in vitro, a few compounds have also been tested both in vitro and in vivo [29]. For instance, E7107 seems to induce profound inhibition of splicing in cellular models [30], while its effects in humans are still under investigation. More recently, cells carrying SF mutations have been found sensitive to treatment with sulfonamides. Leukemia stem cells showed sensitivity to treatment with the aryl sulfonamides (i.e., indisulam) and this sensitivity correlated with increased DDB1 and CUL4 associated factor 15 (DCAF15) expression levels. Therefore, indisulam [N-(3-chloro-7-indolyl)-1,4-benzenedisulfonamide] and other sulfonamides have been investigated for their anticancer activity and demonstrated to drive the recruitment of RNA binding motif protein 39 (RBM39) to DCAF15 for degradation, leading to abnormal mRNA splicing changes (intron retention, exon skipping) [31][32][33]. These observations indicate that cells expressing high levels of DCAF15 might be more susceptible to aryl sulfonamides in general. Another example of the application of small molecules is the anti-proliferative effect of E7070 in combination with idarubicin and cytarabine, which is being investigated in relapsed AML and high-risk MDS (NCT01692197). Moreover, also taking into consideration that protein arginine methyltransferases (PRMTs) inhibitors (MS023, GSK591) were found to influence the growth of SRSF2 mutant cells, the clinical activity of the PRMT5, GSK3326595, is currently explored in combination with azacitidine in newly-diagnosed MDS and AML (NCT03614728).
References
- Chen, S.; Benbarche, S.; Abdel-Wahab, O. Splicing factor mutations in hematologic malignancies. Blood 2021, 138, 599–612.
- Bonnal, S.C.; Lopez-Oreja, I.; Valcarcel, J. Roles and mechanisms of alternative splicing in cancer-implications for care. Nat. Rev. Clin. Oncol. 2020, 17, 457–474.
- Zhang, Y.; Qian, J.; Gu, C.; Yang, Y. Alternative splicing and cancer: A systematic review. Signal Transduct. Target. Ther. 2021, 6, 78.
- Trinh, B.Q.; Ummarino, S.; Zhang, Y.; Ebralidze, A.K.; Bassal, M.A.; Nguyen, T.M.; Heller, G.; Coffey, R.; Tenen, D.E.; van der Kouwe, E.; et al. Myeloid lncRNA LOUP mediates opposing regulatory effects of RUNX1 and RUNX1-ETO in t(8;21) AML. Blood 2021, 138, 1331–1344.
- Belluti, S.; Rigillo, G.; Imbriano, C. Transcription factors in cancer: When alternative splicing determines opposite cell fates. Cells 2020, 9, 760.
- Anufrieva, K.S.; Shender, V.O.; Arapidi, G.P.; Pavlyukov, M.S.; Shakhparonov, M.I.; Shnaider, P.V.; Butenko, I.O.; Lagarkova, M.A.; Govorun, V.M. Therapy-induced stress response is associated with downregulation of pre-mRNA splicing in cancer cells. Genome Med. 2018, 10, 49.
- David, J.K.; Maden, S.K.; Weeder, B.R.; Thompson, R.F.; Nellore, A. Putatively cancer-specific exon-exon junctions are shared across patients and present in developmental and other non-cancer cells. NAR Cancer 2020, 2, zcaa001.
- Kahles, A.; Ong, C.S.; Zhong, Y.; Ratsch, G. SplAdder: Identification, quantification and testing of alternative splicing events from RNA-Seq data. Bioinformatics 2016, 32, 1840–1847.
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research Network; et al. Comprehensive analysis of alternative splicing across tumors from 8705 patients. Cancer Cell 2018, 34, 211–224.
- GTEx Consortium; Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group; Statistical Methods groups—Analysis Working Group; Enhancing GTEx (eGTEx) Groups; NIH Common Fund; NIH/NCI; NIH/NHGRI; NIH/NIMH; NIH/NIDA; Biospecimen Collection Source Site—NDRI; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213.
- Saha, A.; Kim, Y.; Gewirtz, A.D.H.; Jo, B.; Gao, C.; McDowell, I.C.; Consortium, G.T.; Engelhardt, B.E.; Battle, A. Co-expression networks reveal the tissue-specific regulation of transcription and splicing. Genome Res. 2017, 27, 1843–1858.
- Shen, L.; Zhang, J.; Lee, H.; Batista, M.T.; Johnston, S.A. RNA transcription and splicing errors as a source of cancer frameshift neoantigens for vaccines. Sci. Rep. 2019, 9, 14184.
- Hershberger, C.E.; Moyer, D.C.; Adema, V.; Kerr, C.M.; Walter, W.; Hutter, S.; Meggendorfer, M.; Baer, C.; Kern, W.; Nadarajah, N.; et al. Complex landscape of alternative splicing in myeloid neoplasms. Leukemia 2020, 35, 1108–1120.
- Biernacki, M.; Foster, K.A.; Clough, C.; Busch, S.; Cummings, C.; Oehler, V.G.; Stirewalt, D.; Doulatov, S.; Bleakley, M. A shared SF3B1 neoantigen is presented on primary malignant cells and induced pluripotent stem cell-derived hematopoietic cells. Blood 2020, 136, 13–14.
- Laumont, C.M.; Vincent, K.; Hesnard, L.; Audemard, E.; Bonneil, E.; Laverdure, J.P.; Gendron, P.; Courcelles, M.; Hardy, M.P.; Cote, C.; et al. Noncoding regions are the main source of targetable tumor-specific antigens. Sci. Transl. Med. 2018, 10, eaau5516.
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep. 2018, 23, 270–281.
- Richters, M.M.; Xia, H.; Campbell, K.M.; Gillanders, W.E.; Griffith, O.L.; Griffith, M. Best practices for bioinformatic characterization of neoantigens for clinical utility. Genome Med. 2019, 11, 56.
- Schischlik, F.; Jager, R.; Rosebrock, F.; Hug, E.; Schuster, M.; Holly, R.; Fuchs, E.; Milosevic Feenstra, J.D.; Bogner, E.; Gisslinger, B.; et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood 2019, 134, 199–210.
- Marty, R.; Kaabinejadian, S.; Rossell, D.; Slifker, M.J.; van de Haar, J.; Engin, H.B.; de Prisco, N.; Ideker, T.; Hildebrand, W.H.; Font-Burgada, J.; et al. MHC-I genotype restricts the oncogenic mutational landscape. Cell 2017, 171, 1272–1283.
- Havens, M.A.; Duelli, D.M.; Hastings, M.L. Targeting RNA splicing for disease therapy. Wiley Interdiscip. Rev. RNA 2013, 4, 247–266.
- Inoue, D.; Chew, G.L.; Liu, B.; Michel, B.C.; Pangallo, J.; D’Avino, A.R.; Hitchman, T.; North, K.; Lee, S.C.; Bitner, L.; et al. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 2019, 574, 432–436.
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol. Ther. 2017, 25, 2075–2092.
- Kierlin-Duncan, M.N.; Sullenger, B.A. Using 5’-PTMs to repair mutant beta-globin transcripts. RNA 2007, 13, 1317–1327.
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869.
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667.
- Shi, X.; Kitano, A.; Jiang, Y.; Luu, V.; Hoegenauer, K.A.; Nakada, D. Clonal expansion and myeloid leukemia progression modeled by multiplex gene editing of murine hematopoietic progenitor cells. Exp. Hematol. 2018, 64, 33–44.
- Buonamici, S.; Yoshimi, A.; Thomas, M.; Seiler, M.; Chan, B.; Caleb, B.; Darmanet, R.; Fekkes, P.; Karr, C.; Keaney, G.F.; et al. H3B-8800, an orally bioavailable modulator of the SF3b complex, shows efficacy in spliceosome-mutant myeloid malignancies. Blood 2016, 128, 966.
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I first-in-human dose escalation study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550.
- Bonnal, S.; Vigevani, L.; Valcarcel, J. The spliceosome as a target of novel antitumour drugs. Nat. Rev. Drug Discov. 2012, 11, 847–859.
- Folco, E.G.; Coil, K.E.; Reed, R. The anti-tumor drug E7107 reveals an essential role for SF3b in remodeling U2 snRNP to expose the branch point-binding region. Genes Dev. 2011, 25, 440–444.
- Han, T.; Goralski, M.; Gaskill, N.; Capota, E.; Kim, J.; Ting, T.C.; Xie, Y.; Williams, N.S.; Nijhawan, D. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356, eaal3755.
- Hsiehchen, D.; Goralski, M.; Kim, J.; Xie, Y.; Nijhawan, D. Biomarkers for RBM39 degradation in acute myeloid leukemia. Leukemia 2020, 34, 1924–1928.
- Wang, E.; Lu, S.X.; Pastore, A.; Chen, X.; Imig, J.; Chun-Wei Lee, S.; Hockemeyer, K.; Ghebrechristos, Y.E.; Yoshimi, A.; Inoue, D.; et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell 2019, 35, 369–384.
More
Information
Subjects:
Allergy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.1K
Revisions:
2 times
(View History)
Update Date:
16 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No