 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Elliott Hall | + 2604 word(s) | 2604 | 2020-08-12 08:22:45 | | | |
| 2 | Elliott Hall | Meta information modification | 2604 | 2020-08-18 19:24:11 | | | | |
| 3 | Rita Xu | Meta information modification | 2604 | 2020-08-19 04:18:13 | | |
Video Upload Options
Epigenetic mechanisms mediate the integration of genetic and environmental factors that are responsible for changes in phenotypes. The organisation of the human genome within three-dimensional space (the 3D genome) is a dynamic epigenetic regulator of phenotypic expression. Mediated through the changing spatial proximity of genomic regions relative to one another in 3D space, ‘chromosome conformations’ have emerged in the past several years as a novel class of molecular switches that regulate cellular and physiological processes. Technological advances in the detection of chromosome conformations have spawned a new class of biomarker - the chromosome conformation signature (CCS) - that identifies chromosomal interactions across multiple genomic loci as a collective marker of distinct epigenomic states. The use of CCSs in basic and clinical research has shown recent applications in identifying disease states, subtyping disease states, and prospectively stratifying individuals according to their likely response to medical intervention.
1. Epigenetics, Chromatin, & Chromosome Conformations
Epigenetic mechanisms involve the integration of DNA-based genetic information with environmental stimuli to enable the coordinated control of gene activity and changes in phenotype without changes in DNA sequence [1][2][3][4]. As such, epigenetic regulation of genes in response to external stimuli is an important modulator of physiological response and adaptation. Indeed, human phenotypes are partially determined by the selective utilisation of an individual’s unique DNA sequence upon exposure to environmental stimuli. However, only around 1–2% of the genome is comprised of DNA sequences for protein-coding genes. The remaining 98–99% of non-protein coding DNA was once considered “junk”, with no known functions. It is now appreciated that much of this non-coding DNA is crucial to the functioning of cells, especially maintaining genome stability and regulating gene activity. While several epigenetic mechanisms act to mediate phenotypes, the structural organization of the genome with the nucleus (the 3D genome) has gained recent appreciation for the critical role it plays in this process.
Within the nucleus, genomic DNA is tightly wrapped around histone proteins in a complex known as chromatin [5]. At this level the functional repeating subunit (146 DNA base pairs (bp)) of chromatin is known as a nucleosome, consisting of eight histone proteins. Nucleosome accessibility regulates the access of transcriptional machinery to DNA, which must unravel before transcription can take place [6]. This unravelling is an epigenetic process known as chromatin remodeling and is regulated by cell-specific histone modifications that mark genes, transcription start sites, and regulatory DNA sequences to control gene expression [5]. Enhancer regions (short stretches of regulatory DNA that can promote or repress gene expression) are often located hundreds or thousands of bp away from transcription start sites [7]. As such, the formation of ‘loops’ in DNA is required to bring regulatory regions into closer physical proximity with promoter regions. The functional organisation of the genome, therefore, is not simply linear along chromosomes, but acts in a dynamic manner in 3D space and makes the spatial organization of the 3D genome a key mediator of epigenetic regulation of gene expression [3][8][9]. How the 3D genome assembles is non-random, highly regulated, and encompasses many sequence-based mediators such as enhancers, super-enhancers, and domain insulators [10]. Thus, analysing changes in 3D genome organisation could provide valuable insight into understanding cellular phenotypes and associated gene expression patterns caused by environmental exposures.
2. Assessment of Chromosome Conformations
Determining the position and organization of chromosomes within the nucleus, and how this positioning changes with respect to environmental stimuli, is critical to understanding the clinical relevance of the 3D genome. Several methods are available to study the 3D genome, with the most well-developed techniques based on chromosome conformation capture (3C) [11][12]. The basis of 3C is to identify and quantify the number of interactions between genomic loci, potentially separated by thousands of nucleotides in the linear genome, within three-dimensional space [13]. Fundamentally, the 3C methodology freezes the 3D genome in time to determine which genomic regions are interacting with each other. Basic 3C involves chemically fixing cells/nuclei with formaldehyde to cross-link DNA and protein interactions, which stabilizes interactions between genomic loci. Next, the genome is fragmented using restriction enzymes, cleaving DNA into fragments at or near specific recognition sites in the genome. Following fragmentation is a proximity-based ligation reaction step under conditions that favour re-ligation between cross-linked interacting fragments that were physically proximal during fixation over re-ligation between fragments that are not cross-linked. The result is a stable 3C ‘library’ of interactions which can be detected and quantified using different approaches such as PCR and next generation sequencing. Several methodological variants based on 3C have been developed to study genome architecture and are summarised elsewhere [12][14][15][16]. Each varies by cost, resolution, bandwidth, level of stochastic background and throughput, and can be used in combination to provide a view of the genome in vivo [12].
3. Chromosome Conformation Signatures (CCSs)
A single interaction between two genomic regions in 3D space is referred to as a “chromosome conformation”. When multiple conformations are assessed concurrently, together with a clinical endpoint or other phenotypic measure, a “chromosome conformation signature” (CCS) is obtained [12]. Recently, an approach with a focus on clinical and industrial scale screening and application using CCSs was developed [17][18][19][20]. Like the original 3C methodology, EpiSwitch™ detects the absence or presence of interactions between two sites in the genome. However, unlike traditional 3C, EpiSwitch leverages on several important advantages. It uses critical functional filtering of stochastic background with its proprietary in silico pattern recognition annotations for potential interaction sites across the whole genome. It is also based on an industrial processing technology that converts chromosome conformation analytes into simple sequence-based analytes using robotic platforms within few hours. This allows for biomarker screening efforts to focus at high resolution on highly reproducible conditional chromosome conformation targets. Another difference between this approach and conventional 3C is scale. While only single interaction pairs at a time are identified by PCR in 3C, EpiSwitchTM screening employs a comparative customized genomic hybridization (CGH) array, with the ability to assess the absence or presence of over 1 million regulatory interaction pairs at once. This initial screening can then be followed by PCR or sequencing-based detection for CCSs of interest.
The dynamic nature of chromatin organisation [21] suggests there may be CCSs associated with both transient responses and more persistent phenotypic profiles. Application of CCSs [17][18] has shown that this biomarker modality can be applied to whole blood samples [19] to provide stable, binary readouts between two states (pre-intervention vs. post-intervention, disease vs. non-diseased) based on the defined validated signature [12][17][20]. Consequently, CCS technologies identify the flexibility or inflexibility of epigenomic states, rather than the magnitude of gene expression, across multiple loci, and can do so on an individual basis. Notably, the reorganisation of CCSs in response to physiological stimuli leading to changes of phenotype appears to be one of the earliest detectable events, preceding several other epigenetic modifications, transcription factor binding and transcription [22]. Due to the fact that DNA is spatially organized into 3D structures and distal genomic regions can be brought into proximity through chromatin folding, it would be expected that such DNA sequences may also exhibit coordinated epigenetic marks, such as histone modifications and DNA methylation. Indeed, a recent study using a variation of CCS, termed Methyl-HiC, revealing coordinated DNA methylation status between distal genomic segments that are in spatial proximity in the nucleus [22]. Such combined approaches will be important to understand how epigenetic marks are dynamically regulated with characteristic patterns in different tissues.
4. Applications of CCSs in Humans
To date, the initial application of CCSs have been used in clinical medicine, where they have been applied to diagnose disease and predict response to therapeutic intervention.
4.1. Disease diagnosis
The earliest examples of applying 3D genomics in clinical medicine have come in disease diagnosis, particularly for diseases where conventional diagnoses are based on invasive procedures or lacked sufficient discriminatory power. A representative example of the process of applying CCS to stratification of human populations is shown in Figure 1.
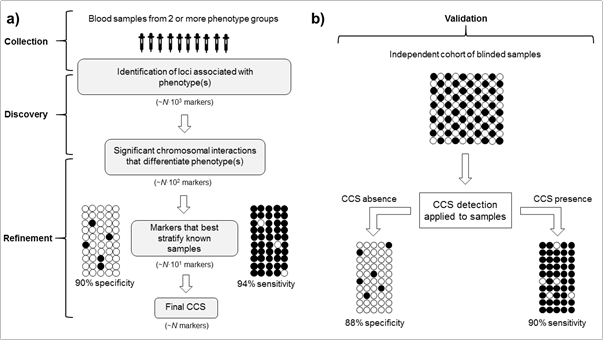
Figure 1. Defining a chromosome conformation signature (CCS) from sample collection to validation. (a) Discovery and refinement. Blood samples from phenotype groups (phenotype A = white dots, phenotype B = black dots) are screened for chromosomal interactions associated with each phenotype, with statistical refinement to determine the best discriminatory markers. Known samples are stratified to determine the sensitivity and specificity of the marker set. Specificity and sensitivity values are hypothetical. Typically, the number of markers in final CCS (N) would be ~5–15. (b) Validation. Blinded, independent samples are used to evaluate whether the final CCS identifies phenotypic groups with accuracy.
A seminal pilot study for clinical 3D genomics came in melanoma, where the limitations of historical biopsy-based diagnostic procedures begot the need for improved methods for early detection and screening. Using a modified 3C assay on small volume blood samples, a fifteen-marker CCS covering five genomic loci with known links to the development of melanoma was identified. The CCS could differentiate patients with melanoma from healthy controls 80% of the time, with 85% sensitivity and 75% specificity [18]. 3D genomic analysis has also been used in neurological diseases, where clinical diagnoses can be difficult and often rely on expensive, invasive, and time-consuming testing. Amyotrophic lateral sclerosis (ALS, or Lou Gherig’s Disease), a degenerative neurologic disorder characterized by progressive muscle weakness, paralysis, and death is one such example. Using an iterative, step-wise approach combining the assessment of chromosome conformations via CGH array and PCR-based screening, a panel of chromosome conformations was developed on a ‘training’ cohort of ALS patients and assessed on a blinded cohort; achieving sensitivity of 88% and specificity of 75% [17]. In a related study in ALS, chromosome conformations have been used to predict probable disease progression course. Taking a similar ‘training’ and ‘assessment’ cohort approach, a longitudinal cross section of ALS patients was used to develop a blood-based CCS that when measured at baseline (after disease diagnosis), could assign newly diagnosed patients with a probable disease course (‘Faster’ or ‘Slower’) based on decline in ALSFRS-R [23]. These findings have been further translated into the clinical trial setting, where the ability of chromosome conformations to predict probable disease course are being assessed by Mitsubishi Tanabe Pharma America in a trial of ALS patients on edaravone (Radicava) therapy [24].
4.2. Therapeutic response prediction
One of the most exciting applications of 3D genomics lies in profiling diseased patients and making predictions on the likelihood of response to a given therapy or class of therapies prior to clinical intervention. Recently, a CCS that could predict inadequate response to methotrexate (MTX), a drug used to treat rheumatoid arthritis (RA), was described in early RA patients [19]. Blood samples taken from early RA patients prior to treatment were used to identify a ‘non-response’ CCS. Using a stepwise approach, a five marker CCS was identified and correctly classified 90% of existing patients as MTX responders or non-responders, with 86% specificity in a blinded test cohort and demonstrating the power of using a CCS-based approach to identify patients’ likelihood of response to medical intervention.
Oncological diseases present perhaps the largest area of clinical medicine where predictors of therapeutic response are needed. In a recent study in diffuse large B-cell lymphoma (DLBCL), a heterogenous blood cancer, a novel approach using 3D genomics was used to develop a molecular disease classifier that could be used to guide therapeutic decision making. DLBCL classification recognizes two main subtypes, germinal center B-cell-like (GCB) and activated B-cell-like (ABC), which have very different clinical courses, along with the unclassified (Type III) subtype. Patients with GCB subtypes generally have more favourable survival outcomes relative to ABC subtypes and this, along with the observation that patients with different subtypes respond differently to therapeutic intervention, are used to make treatment decisions in newly diagnosed patients. As such, having a reliable assay to determine DLBCL subtypes has important implications in guiding the clinical approach to the use of existing therapies, as well as in the development of new drugs. Recently, the development of a CCS for accurately subtyping DLBCL was published by Roche/Genentech offering a robust, complementary method for non-invasive DLBCL prognostic stratification across all three subtypes: ABC/GCB and Type III from whole blood samples using CCSs readouts [20]. The study was one of the first to highlight several attractive features of 3D genomics as a clinical biomarker modality in oncology including the high biochemical stability of the markers, the ability to provide readouts from very small amounts of blood, and the rapid turnaround time compared to gold standard gene expression-based approaches. Future applications of this complementary assay are envisioned to enable prospective selection of patients for therapeutic clinical trials for new drugs to treat DLBCL and ultimately, as a guide to help physicians make patient management decisions in clinical practice.
Targeted cancer therapy has emerged in the past few years as one of the most effective therapeutic strategies for a diverse set of oncological conditions, from solid tumours to blood-based cancers. Within targeted cancer therapy, immune checkpoint inhibitor (CPI) therapy has risen to particular prominence, in large part to some key early demonstrations of clinical success of drugs like Keytruda (pembrolizumab), an antibody targeting the immune checkpoint protein PD-L1. Following the initial approval of pembrolizumab for melanoma and non-small cell lung cancer (NSCLC) in 2014, the drug has seen subsequent approvals in several cancer indications. However, despite the multiple approvals for pembrolizumab and other CPI drugs like it; a majority of patients will not respond to CPI therapy and predicting who will respond to a given CPI remains a major clinical challenge. In a study presented by EMD Serono at the 2019 Society for the Immunotherapy of Cancer (SITC) conference, the first approaches using 3D genomics as a molecular tool to predict response to CPI therapy in non-small cell lung cancer (NSCLC) were reported. The CCS achieved high accuracy (84%), positive predictive value (PPV) (0.87) and negative predictive value (NPV) (0.81) for patients undergoing pembrolizumab as a second line treatment for NSCLC in the test set [25]. Altogether, the CCS study encompasses analysis of patients with NSCLC or melanoma treated with avelumab (an anti–PD-L1 antibody) or pembrolizumab [25]. A CCS marker panel, trained using baseline samples from patients receiving different therapies, achieved good generalized performance in prediction of response across two test sets where avelumab was used as a therapy. In addition, the marker panel retained good PPV (>0.7) when identifying responders to pembrolizumab. Last, when a multivariable survival model was run assessing a variety of clinical and molecular variables, the CCS was one of the only statistically significant predictors of response, suggesting independent power of the approach.
4.3. Human health
While the initial applications of 3D genomic assessments using CCSs have been in the context of human disease, their use in non-diseased subjects presents an exciting new field of application. The heightened social awareness of the importance of maintaining a healthy lifestyle coupled with increasing scrutiny of the environmental, dietary, and lifestyle factors that influence human health and well-being make this a prime area for novel molecular tools to assess the broader phenotype of ‘health’. Indeed, the first set of prospective studies using CCS as a predictor of response to exercise/training has recently been published [26]. When coupled with the rapid advancement in ‘wearable’ digital health technology, the coming years should see exciting advances in the field of health epigenetics.
Together, the existing applications of CCSs demonstrate an ability to efficiently stratify samples from individuals, signifying the potential value of CCS-based approaches in the pursuit of personalised approaches across disciplines.
5. Conclusion
The 3D genome is a major regulator of human phenotypic expression. The development of new molecular tools to assess dynamic changes in genomic organization have opened up exciting new avenues of research and applied use in humans. Promising early studies in the fields of oncology and immune-driven disease are indicators that the use of CCSs as diagnostic and prognostic approaches have a bright future ahead.
References
- McGee, S.L.; Walder, K.R.; Exercise and the Skeletal Muscle Epigenome.. Cold Spring Harb. Perspect. Med. 2017, 7, a029876..
- Henikoff, S.; Greally, J.M.; Epigenetics, cellular memory and gene regulation.. Curr. Biol. 2016, 26, R644-R648.
- Ramani, V.; Shendure, J.; Duan, Z.; Understanding Spatial Genome Organization: Methods and Insights. . Genom. Proteom. Bioinform. 2016, 14, 7-20.
- Alyamani, R.A.S.; Murgatroyd, C.; Epigenetic Programming by Early-Life Stress. . Prog. Mol. Biol. Transl. Sci. 2018, 157, 133-150.
- Dunham, I.; Kundaje, A.; Aldred, S.F.; Collins, P.J.; Davis, C.A.; Doyle, F.; Epstein, C.B.; Frietze, S.; Harrow, J.; Kaul, R.; et al.et al. An integrated encyclopedia of DNA elements in the human genome.. Nature 2012, 489, 57-74.
- Van Holde, K.E. . Chromatin; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; pp. ..
- Sanyal, A.; Lajoie, B.R.; Jain, G.; Dekker, J.; The long-range interaction landscape of gene promoters.. Nature 2012, 489, 109-113.
- Fraser, P.; Bickmore, W.; Nuclear organization of the genome and the potential for gene regulation. . Nature 2007, 447, 413-417.
- West, A.G.; Fraser, P.; Remote control of gene transcription.. Hum. Mol. Genet. 2005, 14, R101-R111.
- Kloetgen, A.; Thandapani, P.; Tsirigos, A.; Aifantis, I.; 3D Chromosomal Landscapes in Hematopoiesis and Immunity. . Trends Immunol. 2019, 40, 809-824.
- de Wit, E.; de Laat, W.; A decade of 3C technologies: Insights into nuclear organization. Genes Dev. 2012, 26, 11-24.
- Crutchley, J.L.; Wang, X.Q.; Ferraiuolo, M.A.; Dostie, J.; Chromatin conformation signatures: Ideal human disease biomarkers? . Biomark. Med. 2010, 4, 611-629.
- Hakim, O.; Misteli, T.; SnapShot: Chromosome confirmation capture.. Cell 2012, 148, 1068.e1–1068.e2.
- Mishra, A.; Hawkins, R.D.; Three-dimensional genome architecture and emerging technologies: Looping in disease. . Genome Med 2017, 9, 87.
- Grob, S.; Cavalli, G.; Technical Review: A Hitchhiker's Guide to Chromosome Conformation Capture. . Methods Mol. Biol. 2018, 1675, 233-246.
- Li, Y.; Tao, T.; Du, L.; Zhu, X.; Three-dimensional genome: Developmental technologies and applications in precision medicine.. J. Hum. Genet. 2020, 65, 497-511.
- Salter, M.; Corfield, E.; Ramadass, A.; Grand, F.; Green, J.; Westra, J.; Lim, C.R.; Farrimond, L.; Feneberg, E.; Scaber, J.; et al.et al. Initial Identification of a Blood-Based Chromosome Conformation Signature for Aiding in the Diagnosis of Amyotrophic Lateral Sclerosis. . EBioMedicine 2018, 33, 169-184.
- Bastonini, E.; Jeznach, M.; Field, M.; Juszczyk, K.; Corfield, E.; Dezfouli, M.; Ahmat, N.; Smith, A.; Womersley, H.; Jordan, P.; et al.et al. Chromatin barcodes as biomarkers for melanoma.. Pigment Cell Melanoma Res. 2014, 27, 788-800.
- Carini, C.; Hunter, E.; Ramadass, A.S.; Green, J.; Akoulitchev, A.; McInnes, I.B.; Goodyear, C.S.; Chromosome conformation signatures define predictive markers of inadequate response to methotrexate in early rheumatoid arthritis.. J. Transl. Med. 2018, 16, 18.
- Hunter, E.; McCord, R.; Ramadass, A.S.; Green, J.; Westra, J.W.; Mundt, K.; Akoulitchev, A.; Comparative molecular cell-of-origin classification of diffuse large B-cell lymphoma based on liquid and tissue biopsies. . Transl. Med. Commun. 2020, 5, 5.
- Hubner, M.R.; Spector, D.L.; Chromatin dynamics.. Annu. Rev. Biophys. 2010, 39, 471-489.
- 22. Christova, R.; Jones, T.; Wu, P.J.; Bolzer, A.; Costa-Pereira, A.P.; Watling, D.; Kerr, L.M.; Sheer, D.; P-STAT1 mediates higher-order chromatin remodelling of the human MHC in response to IFNgamma. J. Cell Sci. 2007, 120, 3362-3270.
- 23. Salter, M.; Elvidge, W.; Ramadass, A.; Womersley, H.; Grand, F.; Green, J.; Kent, L.; Ossher, L.; Turner, M.; Talbot, K.; Cudkowicz, M. Epigenetic signatures and early detection of neurodegenerative diseases. Lancet Neurology Conference, 2016.
- Oxford Biodynamics Joins ALS Biomarker Study Sponsored by Mitsubishi Tanabe Pharma America . Mitsubishi Tanabe Pharma America. Retrieved 2020-8-18
- Shah, P; Hunter, E.; Potluri, S.; Zhang, S.; Dezfouli, M.; Back, J.; James, L.; Jandor, N.; Powell, R.; Salter, M.; et al.Ramadass, A.Green, J.Westra, W.Dong, H.Dronca, R.Markovic, S.Akoulitchev, A.Cai, T.Robbins, P. Development and validation of baseline predictive biomarkers for response to avelumab n second -line (2L) non-small cell lung cancer (NSCLC) using EpiSwitch epigenetic profiling.. J Immunother Cancer 2019, 7, 78-9.
- Hall, E.C.R.; Murgatroyd, C.; Stebbings, G.K; Cunniffe, B.; Harle, L.; Salter, M.; Ramadass, A.; Westra, J.W.; Hunter, E.; Akoulitchev, A.; et al.Williams, A.G The Prospective Study of Epigenetic Regulatory Profiles in Sport and Exercise Monitored Through Chromosome Conformation Signatures. Genes 2020, 11, 905.

