 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yuanxin Zhao | + 2129 word(s) | 2129 | 2021-12-06 03:05:52 | | | |
| 2 | Conner Chen | Meta information modification | 2129 | 2021-12-17 04:37:15 | | | | |
| 3 | Conner Chen | Meta information modification | 2129 | 2021-12-17 04:37:30 | | |
Video Upload Options
One of the most striking hallmarks shared by various neurodegenerative diseases, including Parkinson’s disease, Alzheimer’s disease and amyotrophic lateral sclerosis, is microglia-mediated and astrocyte-mediated neuroinflammation. Although inhibitions of both harmful proteins and aggregation are major treatments for neurodegenerative diseases, whether the phenomenon of non-normal protein or peptide aggregation is causally related to neuronal loss and synaptic damage is still controversial. Currently, excessive production of reactive oxygen species (ROS), which induces mitochondrial dysfunction in neurons that may play a key role in the regulation of immune cells, is proposed as a regulator in neurological disorders. In this review, we propose that mitochondrial DNA (mtDNA) release due to ROS may act on microglia and astrocytes adjacent to neurons to induce inflammation through activation of innate immune responses (such as cGAS/STING). Elucidating the relationship between mtDNA and the formation of a pro-inflammatory microenvironment could contribute to a better understanding of the mechanism of crosstalk between neuronal and peripheral immune cells and lead to the development of novel therapeutic approaches to neurodegenerative diseases.
1. Possible Mechanisms of mtDNA Release
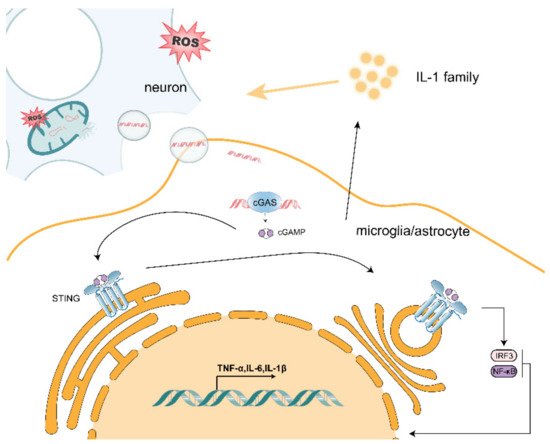
2. Effect of mtDNA on Microglia
3. Effect of mtDNA on Astrocytes
4. mtDNA Promotes Activation of the cGAS/STING Pathway
5. mtDNA/cGAS/STING Pathway Exacerbates Neurodegenerative Disease
References
- Krysko, D.; Agostinis, P.; Krysko, O.; Garg, A.; Bachert, C.; Lambrecht, B.N.; Vandenabeele, P. Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol. 2011, 32, 157–164.
- Zhao, M.; Wang, Y.; Li, L.; Liu, S.; Wang, C.; Yuan, Y.; Yang, G.; Chen, Y.; Cheng, J.; Lu, Y.; et al. Mitochondrial ROS promote mitochondrial dysfunction and inflammation in ischemic acute kidney injury by disrupting TFAM-mediated mtDNA maintenance. Theranostics 2021, 11, 1845–1863.
- Cheng, A.N.; Cheng, L.-C.; Kuo, C.-L.; Lo, Y.K.; Chou, H.-Y.; Chen, C.-H.; Wang, Y.-H.; Chuang, T.-H.; Cheng, S.-J.; Lee, A.Y.-L. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1–mediated immunoescape via STING-IFN signaling and extracellular vesicles. J. Immunother. Cancer 2020, 8, e001372.
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536.
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. 2018, 37, e99238.
- Huang, L.S.; Hong, Z.; Wu, W.; Xiong, S.; Gao, X.; Rehman, J.; Malik, A.B. mtDNA Activates cGAS Signaling and Suppresses the YAP-Mediated Endothelial Cell Proliferation Program to Promote Inflammatory Injury. Immunity 2020, 52, 475–486.e5.
- Bao, D.; Zhao, J.; Zhou, X.; Yang, Q.; Chen, Y.; Zhu, J.; Yuan, P.; Yang, J.; Qin, T.; Wan, S.; et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene 2019, 38, 5007–5020.
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230.
- Li, T.; Tan, X.; Li, S.; Al-Nusaif, M.; Le, W. Role of Glia-Derived Extracellular Vesicles in Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 765395.
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075.
- Torralba, D.; Baixauli, F.; Villarroya-Beltri, C.; Fernández-Delgado, I.; Latorre-Pellicer, A.; Acín-Pérez, R.; Martín-Cófreces, N.B.; Jaso-Tamame, Á.L.; Iborra, S.; Jorge, I.; et al. Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through anti-gen-driven contacts. Nat. Commun. 2018, 9, 2658.
- Gambardella, S.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Campopiano, R.; Centonze, D.; Fornai, F. ccf-mtDNA as a Potential Link Between the Brain and Immune System in Neuro-Immunological Disorders. Front. Immunol. 2019, 10, 1064.
- Liao, Y.; Cheng, J.; Kong, X.; Li, S.; Li, X.; Zhang, M.; Zhang, H.; Yang, T.; Dong, Y.; Li, J.; et al. HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 2020, 10, 9644–9662.
- Mathur, V.; Burai, R.; Vest, R.T.; Bonanno, L.; Lehallier, B.; Zardeneta, M.E.; Mistry, K.N.; Do, D.; Marsh, S.; Abud, E.M.; et al. Activation of the STING-Dependent Type I Interferon Response Reduces Microglial Reactivity and Neuroinflammation. Neuron 2017, 96, 1290–1302.e6.
- Tsilioni, I.; Theoharides, T.C. Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1β. J. Neuroinflamm. 2018, 15, 1–8.
- Nakanishi, H.; Hayashi, Y.; Wu, Z. The role of microglial mtDNA damage in age-dependent prolonged LPS-induced sickness behavior. Neuron Glia Biol. 2011, 7, 17–23.
- Nasi, M.; De Gaetano, A.; Bianchini, E.; De Biasi, S.; Gibellini, L.; Neroni, A.; Mattioli, M.; Pinti, M.; Tartaro, D.L.; Borella, R.; et al. Mitochondrial damage-associated molecular patterns stimulate reactive oxygen species production in human microglia. Mol. Cell. Neurosci. 2020, 108, 103538.
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142.
- Yu, C.-H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18.
- Lee, S.; Kim, S.; Kang, H.Y.; Lim, H.R.; Kwon, Y.; Jo, M.; Jeon, Y.M.; Kim, S.R.; Kim, K.; Ha, C.M.; et al. The overexpression of TDP-43 in astrocytes causes neurodegeneration via a PTP1B-mediated inflammatory re-sponse. J. Neuroinflamm. 2020, 17, 299.
- Hu, J.; Bibli, S.I.; Wittig, J.; Zukunft, S.; Lin, J.; Hammes, H.-P.; Popp, R.; Fleming, I. Soluble epoxide hydrolase promotes astrocyte survival in retinopathy of prematurity. J. Clin. Investig. 2019, 129, 5204–5218.
- Ignatenko, O.; Chilov, D.; Paetau, I.; De Miguel, E.; Jackson, C.B.; Capin, G.; Paetau, A.; Terzioglu, M.; Euro, L.; Suomalainen, A. Loss of mtDNA activates astrocytes and leads to spongiotic encephalopathy. Nat. Commun. 2018, 9, 1–12.
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674.
- Zhang, X.; Wu, J.; Liu, Q.; Li, X.; Li, S.; Chen, J.; Hong, Z.; Wu, X.; Zhao, Y.; Ren, J. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis. 2020, 11, 1050.
- E Leurs, C.; Podlesniy, P.; Trullas, R.; Balk, L.; Steenwijk, M.D.; Malekzadeh, A.; Piehl, F.; Uitdehaag, B.M.; Killestein, J.; Van Horssen, J.; et al. Cerebrospinal fluid mtDNA concentration is elevated in multiple sclerosis disease and responds to treatment. Mult. Scler. J. 2017, 24, 472–480.
- Podlesniy, P.; Figueiro-Silva, J.; Llado, A.; Antonell, A.; Sanchez-Valle, R.; Alcolea, D.; Lleo, A.; Molinuevo, J.L.; Serra, N.; Trullas, R. Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann. Neurol. 2013, 74, 655–668.
- Cervera-Carles, L.; Alcolea, D.; Estanga, A.; Ecay-Torres, M.; Izagirre, A.; Clerigué, M.; Garcia-Sebastian, M.; Villanúa, J.; Escalas, C.; Blesa, R.; et al. Cerebrospinal fluid mitochondrial DNA in the Alzheimer’s disease continuum. Neurobiol. Aging 2017, 53, 192.e1–192.e4.
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262.
- Brambilla, G.; Gallotti, C. . Radiol. Med. 1987, 74, 49–58.
- Jauhari, A.; Baranov, S.V.; Suofu, Y.; Kim, J.; Singh, T.; Yablonska, S.; Li, F.; Wang, X.; Oberly, P.; Minnigh, M.B.; et al. Melatonin inhibits cytosolic mitochondrial DNA–induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J. Clin. Investig. 2021, 131.

