 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mohammad Asif Khan | + 3032 word(s) | 3032 | 2021-09-10 16:30:04 | | | |
| 2 | Peter Tang | Meta information modification | 3032 | 2021-12-14 06:43:45 | | |
Video Upload Options
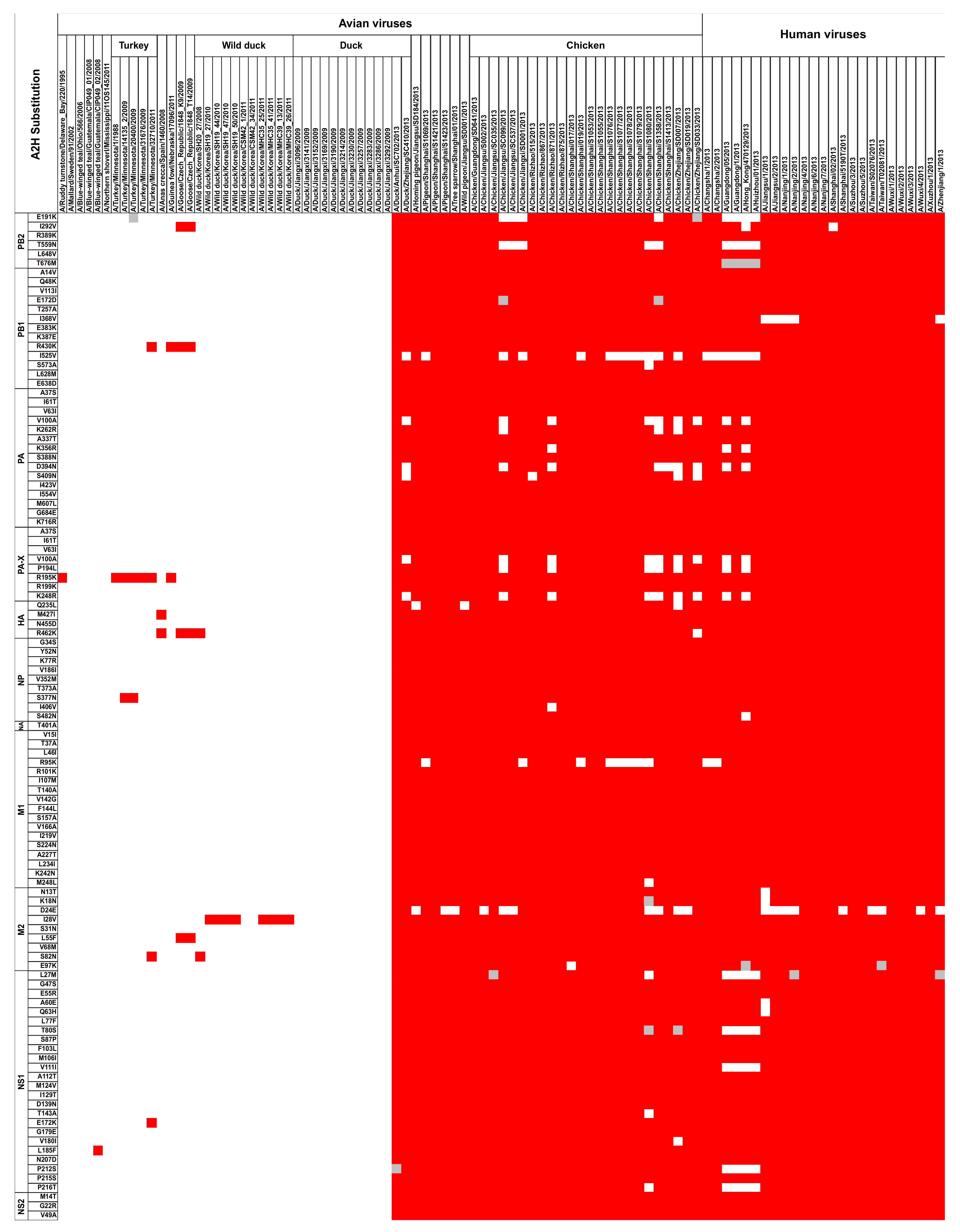
Avian influenza virus A (H7N9), after circulating in avian hosts for decades, was identified as a human pathogen in 2013. Herein, we focus on the quantification of the virus diversity and the identification of amino acid substitutions that are possibly essential for human adaptation.
1. Introduction
2. Current Insight on H7N9 Virus and Adaptation to Human Hosts
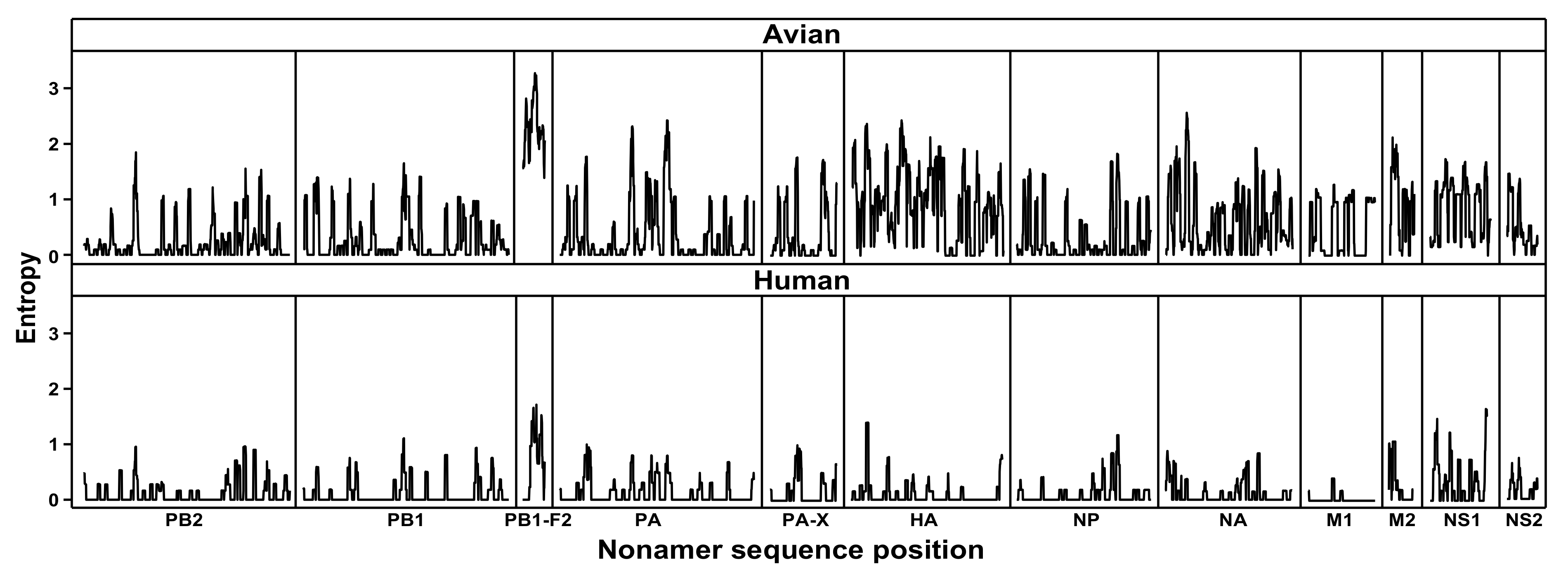
Figure 1. Protein sequence diversity of avian and human influenza A (H7N9) viruses. Shannon’s entropy was used as a general measure of protein sequence diversity for each aligned nonamer (nine amino acids) position of the H7N9 avian (upper) and human (lower) virus proteomes. The entropy values indicate the level of variability at the corresponding nonamer positions, with a zero representing completely conserved sites and high entropy values of about 3 or higher marking highly variable sites.
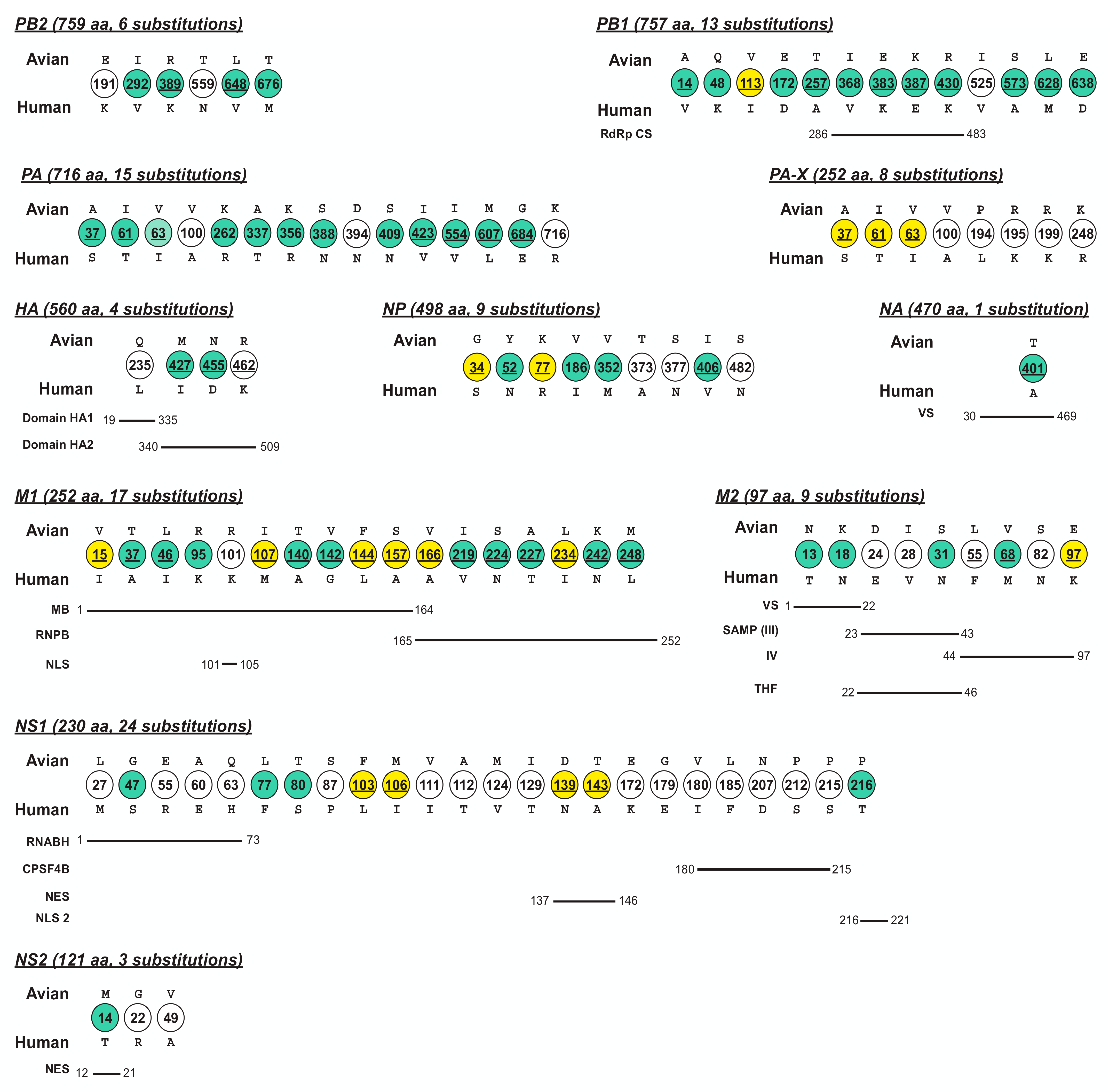
3. Conclusion
References
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53.
- Taubenberger, J.K.; Kash, J.C. Influenza virus evolution, host adaptation, and pandemic formation. Cell Host Microbe 2010, 7, 440–451.
- Nowak, M.A. What is a quasispecies? Trends Ecol. Evol. 1992, 7, 118–121.
- Steinhauer, D.A. Influenza: Pathways to human adaptation. Nature 2013, 499, 412–413.
- Fitch, W.M.; Leiter, J.M.; Li, X.Q.; Palese, P. Positive Darwinian evolution in human influenza A viruses. Proc. Natl. Acad. Sci. USA 1991, 88, 4270–4274.
- Uyeki, T.M.; Peiris, M. Novel Avian Influenza A Virus Infections of Humans. Infect. Dis. Clin. North Am. 2019, 33, 907–932.
- Wendel, I.; Matrosovich, M.; Klenk, H.D. SnapShot: Evolution of human influenza A viruses. Cell Host Microbe 2015, 17, 416.e1.
- Morens, D.M.; Fauci, A.S. The 1918 influenza pandemic: Insights for the 21st century. J. Infect. Dis. 2007, 195, 1018–1028.
- Potter, C.W. A history of influenza. J. Appl. Microbiol. 2001, 91, 572–579.
- Xu, Y.; Peng, R.; Zhang, W.; Qi, J.; Song, H.; Liu, S.; Wang, H.; Wang, M.; Xiao, H.; Fu, L.; et al. Avian-to-Human Receptor-Binding Adaptation of Avian H7N9 Influenza Virus Hemagglutinin. Cell Rep. 2019, 29, 2217–2228.e5.
- Poovorawan, Y.; Pyungporn, S.; Prachayangprecha, S.; Makkoch, J. Global alert to avian influenza virus infection: From H5N1 to H7N9. Pathog. Glob. Health 2013, 107, 217–223.
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N. Engl. J. Med. 2013, 368, 1888–1897.
- Lam, T.T.-Y.; Wang, J.; Shen, Y.; Zhou, B.; Duan, L.; Cheung, C.-L.; Ma, C.; Lycett, S.J.; Leung, C.Y.-H.; Chen, X.; et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 2013, 502, 241–244.
- Zhou, J.; Wang, D.; Gao, R.; Zhao, B.; Song, J.; Qi, X.; Zhang, Y.; Shi, Y.; Yang, L.; Zhu, W.; et al. Biological features of novel avian influenza A (H7N9) virus. Nature 2013, 499, 500–503.
- Watanabe, T.; Kiso, M.; Fukuyama, S.; Nakajima, N.; Imai, M.; Yamada, S.; Murakami, S.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Sakoda, Y.; et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 2013, 501, 551–555.
- Neumann, G.; Macken, C.A.; Kawaoka, Y. Identification of amino acid changes that may have been critical for the genesis of A(H7N9) influenza viruses. J. Virol. 2014, 88, 4877–4896.
- Lam, T.T.-Y.; Zhou, B.; Wang, J.; Chai, Y.; Shen, Y.; Chen, X.; Ma, C.; Hong, W.; Chen, Y.; Zhang, Y.; et al. Dissemination, divergence and establishment of H7N9 influenza viruses in China. Nature 2015, 522, 102–105.
- Yang, Z.F.; Mok, C.K.P.; Liu, X.Q.; Li, X.B.; He, J.F.; Da Guan, W.; Xu, Y.H.; Pan, W.Q.; Chen, L.Y.; Lin, Y.P.; et al. Clinical, virological and immunological features from patients infected with re-emergent avian-origin human H7N9 influenza disease of varying severity in Guangdong province. Sci. Transl. Med. 2015, 7, e0117846.
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. Pandemic Influenza Viruses-Hoping for the Road Not Taken. N. Engl. J. Med. 2013, 368, 1–4.
- Uyeki, T.M.; Cox, N.J. Global Concerns Regarding Novel Influenza A (H7N9) Virus Infections. N. Engl. J. Med. 2013, 368, 1–3.
- Cui, L.; Liu, D.; Shi, W.; Pan, J.; Qi, X.; Li, X.; Guo, X.; Zhou, M.; Li, W.; Li, J.; et al. Dynamic reassortments and genetic heterogeneity of the human-infecting influenza A (H7N9) virus. Nat. Commun. 2014, 5, 3142.
- Watanabe, T.; Watanabe, S.; Maher, E.A.; Neumann, G.; Kawaoka, Y. Pandemic potential of avian influenza A (H7N9) viruses. Trends Microbiol. 2014, 22, 623–631.
- Neumann, G.; Kawaoka, Y. Transmission of influenza A viruses. Virology 2015, 479–480, 234–246.
- CDC. Asian Lineage Avian Influenza A (H7N9) Virus. Centers Dis. Control Prev. 2018.
- Bisset, A.T.; Hoyne, G.F. Evolution and Adaptation of the Avian H7N9 Virus into the Human Host. Microorganisms 2020, 8, 778.
- Chen, L.; Sun, L.; Li, R.; Chen, Y.; Zhang, Z.; Xiong, C.; Zhao, G.; Jiang, Q. Is a highly pathogenic avian influenza virus H5N1 fragment recombined in PB1 the key for the epidemic of the novel AIV H7N9 in China, 2013? Int. J. Infect. Dis. 2016, 43, 85–89.
- De Jong, R.M.C.; Stockhofe-Zurwieden, N.; Verheij, E.S.; de Boer-Luijtze, E.A.; Ruiter, S.J.M.; de Leeuw, O.S.; Cornelissen, L.A.H.M. Rapid emergence of a virulent PB2 E627K variant during adaptation of highly pathogenic avian influenza H7N7 virus to mice. Virol. J. 2013, 10, 1–11.
- Mok, C.K.P.; Lee, H.H.Y.; Lestra, M.; Nicholls, J.M.; Chan, M.C.W.; Sia, S.F.; Zhu, H.; Poon, L.L.M.; Guan, Y.; Peiris, J.S.M. Amino acid substitutions in polymerase basic protein 2 gene contribute to the pathogenicity of the novel A/H7N9 influenza virus in mammalian hosts. J. Virol. 2014, 88, 3568–3576.
- Zhang, H.; Li, X.; Guo, J.; Li, L.; Chang, C.; Li, Y.; Bian, C.; Xu, K.; Chen, H.; Sun, B. The PB2 E627K mutation contributes to the high polymerase activity and enhanced replication of H7N9 influenza virus. J. Gen. Virol. 2014, 95, 779–786.
- Dortmans, J.C.F.M.; Dekkers, J.; Wickramasinghe, I.N.A.; Verheije, M.H.; Rottier, P.J.M.; van Kuppeveld, F.J.M.; de Vries, E.; de Haan, C.A.M. Adaptation of novel H7N9 influenza A virus to human receptors. Sci. Rep. 2013, 3, 3058.
- Tharakaraman, K.; Jayaraman, A.; Raman, R.; Viswanathan, K.; Stebbins, N.W.; Johnson, D.; Shriver, Z.; Sasisekharan, V.; Sasisekharan, R. Glycan receptor binding of the influenza A Virus H7N9 hemagglutinin. Cell 2013, 153, 1486–1493.
- Miotto, O.; Heiny, A.T.; Tan, T.W.; August, J.T.; Brusic, V. Identification of human-to-human transmissibility factors in PB2 proteins of influenza A by large-scale mutual information analysis. BMC Bioinformatics 2008, 9 (Suppl. S1), 1–18.
- Miotto, O.; Heiny, A.T.; Albrecht, R.; García-Sastre, A.; Tan, T.W.; August, J.T.; Brusic, V. Complete-proteome mapping of human influenza A adaptive mutations: Implications for human transmissibility of zoonotic strains. PLoS ONE 2010, 5, e9025.
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423. [CrossRef]
- Heiny, A.T.; Miotto, O.; Srinivasan, K.N.; Khan, A.M.; Zhang, G.L.; Brusic, V.; Tan, T.W.; August, J.T. Evolutionarily conserved protein sequences of influenza a viruses, avian and human, as vaccine targets. PLoS ONE 2007, 2, e1190.
- Khan, A.M.; Miotto, O.; Nascimento, E.J.M.; Srinivasan, K.N.; Heiny, A.T.; Zhang, G.L.; Marques, E.T.; Tan, T.W.; Brusic, V.; Salmon, J.; et al. Conservation and variability of dengue virus proteins: Implications for vaccine design. PLoS Negl. Trop. Dis. 2008, 2, e272.
- De Graaf, M.; Fouchier, R.A.M. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 2014, 33, 823–841.
- Xiang, N.; Li, X.; Ren, R.; Wang, D.; Zhou, S.; Greene, C.M.; Song, Y.; Zhou, L.; Yang, L.; Davis, C.T.; et al. Assessing Change in Avian Influenza A(H7N9) Virus Infections During the Fourth Epidemic—China, September 2015–August 2016. Morb. Mortal. Wkly. Rep. 2016, 65, 1390–1394.
- Wang, D.; Yang, L.; Zhu, W.; Zhang, Y.; Zou, S.; Bo, H.; Gao, R.; Dong, J.; Huang, W.; Guo, J.; et al. Two Outbreak Sources of Influenza A (H7N9) Viruses Have Been Established in China. J. Virol. 2016, 90, 5561–5573.
- Arzt, S.; Petit, I.; Burmeister, W.P.; Ruigrok, R.W.H.; Baudin, F. Structure of a knockout mutant of influenza virus M1 protein that has altered activities in membrane binding, oligomerisation and binding to NEP (NS2). Virus Res. 2004, 99, 115–119.
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179.
- Aragón, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortín, J.; Nieto, A. Eukaryotic Translation Initiation Factor 4GI Is a Cellular Target for NS1 Protein, a Translational Activator of Influenza Virus. Mol. Cell. Biol. 2000, 20, 6259–6268.
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; García-Sastre, A. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573.
- Yewdell, J.; García-Sastre, A. Influenza virus still surprises. Curr. Opin. Microbiol. 2002, 5, 414–418.
- Du, W.; Guo, H.; Nijman, V.S.; Doedt, J.; van der Vries, E.; van der Lee, J.; Li, Z.; Boons, G.J.; van Kuppeveld, F.J.M.; de Vries, E.; et al. The 2nd sialic acid-binding site of influenza a virus neuraminidase is an important determinant of the hemagglutinin-neuraminidase-receptor balance. PLoS Pathog. 2019, 15, e1007860.
- Dai, M.; McBride, R.; Dortmans, J.C.F.M.; Peng, W.; Bakkers, M.J.G.; de Groot, R.J.; van Kuppeveld, F.J.M.; Paulson, J.C.; de Vries, E.; de Haan, C.A.M. Mutation of the Second Sialic Acid-Binding Site, Resulting in Reduced Neuraminidase Activity, Preceded the Emergence of H7N9 Influenza A Virus. J. Virol. 2017, 91.
- Kile, J.C.; Ren, R.; Liu, L.; Greene, C.M.; Roguski, K.; Iuliano, A.D.; Jang, Y.; Jones, J.; Thor, S.; Song, Y.; et al. Update: Increase in Human Infections with Novel Asian Lineage Avian Influenza A(H7N9) Viruses During the Fifth Epidemic—China, October 1, 2016–August 7, 2017. MMWR. Morb. Mortal. Wkly. Rep. 2017, 66, 928–932.

