Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jakub Sinsky | + 2149 word(s) | 2149 | 2021-08-31 12:06:25 | | | |
| 2 | Camila Xu | Meta information modification | 2149 | 2021-12-15 02:27:27 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Sinsky, J. Tau protein Interaction Partners. Encyclopedia. Available online: https://encyclopedia.pub/entry/17043 (accessed on 26 July 2026).
Sinsky J. Tau protein Interaction Partners. Encyclopedia. Available at: https://encyclopedia.pub/entry/17043. Accessed July 26, 2026.
Sinsky, Jakub. "Tau protein Interaction Partners" Encyclopedia, https://encyclopedia.pub/entry/17043 (accessed July 26, 2026).
Sinsky, J. (2021, December 13). Tau protein Interaction Partners. In Encyclopedia. https://encyclopedia.pub/entry/17043
Sinsky, Jakub. "Tau protein Interaction Partners." Encyclopedia. Web. 13 December, 2021.
Copy Citation
Tau protein belongs to the family of microtubule-associated proteins (MAPs) and can influence axonal transport and growth, neuronal polarization, and thus the normal function of neurons and the brain.
tau protein
interaction partners
Alzheimer’s disease
tauopathies
1. Introduction
Proteins are essential macromolecules that play important roles in almost any cellular process. They usually do not function alone but rather as complexes with other molecules, mainly with proteins. Protein-protein interactions (PPIs) are elementary for many processes and it is proposed that their dysfunction or deregulation is located upstream, leading to various pathological conditions [1]. In Alzheimer’s disease and other tauopathies, tau protein undergoes pathological modifications that lead to the formation of paired helical filaments (PHF) and neurofibrillary tangles (NT) which belong to the main hallmarks of these diseases. The conversion of physiological tau into its pathological forms and their participation in disease etiology have not been fully understood yet and are still under investigation. In physiological conditions, tau interacts with many protein partners which maintain their proper structure and function. Under pathological conditions, proteins interacting with tau can also participate in its non-physiological modifications leading to the development of neurodegenerative diseases. Interaction partners of tau protein and involved molecular pathways can either initiate and drive the tau pathology or can have neuroprotective roles, by reducing pathological tau changes or inflammation.
Tau protein belongs to the family of microtubule-associated proteins (MAPs) [2][3] and can influence axonal transport and growth [4], neuronal polarization [5], and thus the normal function of neurons and the brain (Figure 1) [6][7].
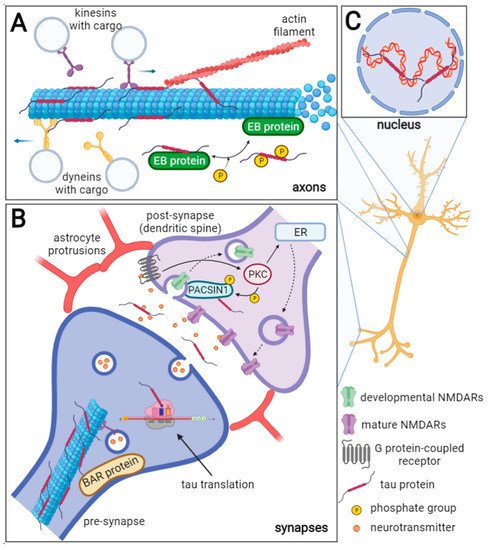
Figure 1. Schematic representation of physiological tau protein functions in neurons. The axonal tau (A) stabilizes microtubules (MTs), and it can also bind actin filaments thus facilitating cytoskeleton networking. Furthermore, tau regulates MT dynamics by interacting with end-binding (EB) proteins. The EB proteins promote and regulate MT nucleation and elongation. Tau inhibits the EB protein binding to MTs and this inhibition is reversed by tau phosphorylation. Tau also competitively inhibits the interaction of dynein and kinesin to MTs and thus influences the intraneuronal transport and cargo release. In the synapses (B), tau protein can be directly translated, and during neuronal activity, it is released into the synaptic cleft. Through linking the MTs and actin filaments, tau can influence synaptic plasticity. Moreover, tau is a known interacting partner of the BAR domain-containing proteins such as BAIAP2, PACSIN1, and BIN1, that ensure the curvature and shaping of the neuronal membrane. Tau may play a role in the removal of developmental NMDARs and their replacement for mature NMDARs in dendrites (dashed arrows) as it is a known substrate of the protein kinase C (PKC), which is activated by the G protein-coupled receptors (GPCR). The PACSIN1 recruits clathrin and dynamin endocytic machinery to the developmental NMDARs and thus mediates their removal. The developmental/mature NMDARs exchange is important for the formation of new synaptic connections. ER—endoplasmic reticulum. In the nucleus (C), tau interacts with DNA and RNA, maintains their integrity, and protects them from oxidative damage. Furthermore, tau may be involved in rRNA-coding DNA transcription and rRNA processing.
In humans, tau protein is expressed mainly in neurons [8], and in lower amounts in oligodendrocytes and astrocytes [9][10][11][12]. Besides the central nervous system (CNS), tau is expressed by peripheral neurons [13], and more recently, tau immunoreactivity was also found in the human submandibular gland and sigmoid colon tissues [14]. Human tau protein is encoded by MAPT gene localized on chromosome 17q21 and consists of 16 exons. In the adult human brain, six tau isoforms are expressed ranging from 352 to 441 amino acids (see Figure 2). Three decades ago, it was proposed that individual tau isoforms may have different functions since they are differently expressed in the fetal and developed brain [15][16][17]. Now, it is known that alternative splicing even varies across neuronal cell types and during neuronal maturation [18][19].
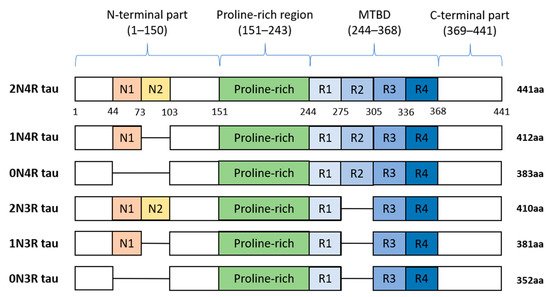
Figure 2. Schematic representation of six human tau protein isoforms and their domains. The N-terminal part is more acidic due to N1 and N2 inserts. The microtubule-binding domain (MTBD) is composed of repeats R1–R4. The number of repeats and N-terminal inserts varies according to the type of tau protein isoform.
Tau protein is distinctly divided into the N-terminal part, proline-rich region (PRR), microtubule-binding domain (MTBD), and C-terminus. The N-terminal domain length is dependent on alternative splicing of exons 2 and 3 which encode acidic amino acids. In MTBD, the splicing-dependent manner of exon 10 results in either 3 (3R) or 4 (4R) microtubule-binding repeats which are essential for binding of tau to individual tubulin heterodimers, and through this interaction tau stabilizes microtubules (MT) [20].
Despite the MT-stabilizing function of tau, its removal or modification had no significant impact on microtubule stability, cellular function, or cognition in mouse models [21][22][23][24][25]. It was shown that the MT-stabilizing function of tau protein is replaceable by microtubule-associated proteins MAP1a [22][26] or MAP1b [5]. However, experiments performed by several other groups on tau-knockout mice revealed that tau can influence the regulation of neuronal activity [27], synaptic plasticity [28], neurogenesis [29], iron export from neurons [30], and long-term depression of synapses [31][32]. Along with other animal disease models where the tau levels were reduced, it was found that depletion of tau protein had a protective effect on neurons against amyloid-beta (Aβ) induced excitotoxicity or by other excitotoxins in mice over-expressing amyloid precursor protein (APP) and presenilin 1 [33][34][35][36][37]. In chemically induced seizure models, hyperexcitability of neurons was reduced in mice with attenuated tau expression [38]. This suggests that endogenous tau is integral for regulating, or rather, upregulating neuronal hyperexcitability in diseased animals. In addition, in mice with impaired function of voltage-dependent sodium and potassium channels, the depletion of tau protein had a protective effect on neurons [39]. These studies suggest that tau protein may play a role in the regulation of neuronal network activity in both pathological and physiological conditions. Moreover, this is also supported by the evidence that tau protein has several additional roles in neurons and the brain [6].
In pathological conditions, the accumulation of insoluble tau aggregates occurs inside neurons, in extracellular space [40][41], and other brain cells such as astrocytes and oligodendrocytes [42][43]. The formation of this stable material is the consequence of abnormally modified and truncated tau proteins which self-aggregate and gradually mature to paired helical filaments (PHF) and neurofibrillary tangles (NFT) which are common hallmarks of several neurodegenerative diseases [44]. The formation of PHFs and NFTs is associated with the engulfment of the cytosol of neurons, failure in intracellular trafficking, and gradual disruption of basic physiological processes that end in apoptosis and neuronal death [45][46][47]. The formation of these aggregates is accompanied by inflammation which on one hand could help in their clearance, but on the other, can exacerbate the pathological processes [48][49][50]. Tau pathology is the main cause of dementia in Alzheimer’s disease and other neurodegenerative diseases, including frontotemporal dementia [51], argyrophilic grain disease [52], corticobasal degeneration [53][54], progressive supranuclear palsy [55], and several other diseases [44]. These disorders, where the accumulation of abnormal tau protein in the brain occurs, are referred to as tauopathies.
2. Roles of Tau Protein in Physiology and Pathology
2.1. Tau and Axonal Transport
Tau protein is enriched in axons where it binds microtubules through its MTBD and participates in their stabilization and regulation. Tau protein’s “free” flanking N-terminal and C-terminal regions interact with various classes of proteins involved in the regulation of cytoskeleton [56][57] and motor proteins kinesins and dyneins [58][59]. Thus, tau participates in the regulation of intraneuronal transport and modulation of microtubule dynamics, which ensures flexible reorganization of cytoskeleton and synaptic transmission. Tau can modulate functions of motor proteins by competitive inhibition of interactions of dynein and kinesin with microtubules, facilitating dynein binding to microtubules, or regulation of transport-vesicle releasing from motor proteins [60][61].
2.2. Tau Protein in Synapses
Besides axons, tau protein occurs also in pre- and post-synapses and in smaller amounts also in dendrites [31][32][33]. Tau protein can be directly translated in synapses and this translation is regulated by synaptic activity [62]. Furthermore, synaptic activity mediates tau protein release into extracellular space including synaptic clefts [63][64][65][66]. When discussing tau in the neuronal synapse, it is important to mention that synapses are complex biological structures also comprising astrocytes. These physiological structures composed of neuronal synapses and astrocyte protrusions are referred to as tripartite synapses [67][68]. Through these protrusions, astrocytes maintain the proper function of synapses through astrocyte–lactate shuttle, glutamate and GABA uptake from the synaptic cleft, growth factors release, and other processes [69]. In tauopathies, tau protein aggregates damage these tripartite synapses and disturb the normal function of neuronal networks [70]. Furthermore, the microglia, which constantly scan the surroundings with their processes, interact regularly with synapses [71][72]. Along with the fact that tau proteins are released during synaptic activity under both, physiological and pathological conditions, the microglia themselves also can mediate the inter-cellular spreading of the tau protein [73]. Released extracellular tau could be capable of interactions with proteins present in the synaptic clefts, thus influencing synaptic functions.
The structure of synapses is determined by the actin filaments, microtubules, and proteins modulating the membrane shape. It was shown that tau can mediate changes in the dendritic cytoskeleton and regulate synaptic plasticity and signaling [74][75]. Tau protein is known to interact with proteins regulating cytoskeleton and membrane curvature which are essential for synapse formation and sustainability [56][76][77][78][79]. Tau binds actin via its PRR, and at the same time also binds microtubules through its MTBD and serves as a crosslinker between microtubules and actin filaments and thus helps to organize the cytoskeleton network [20][80][81]. Furthermore, tau regulates the function of synaptic and extrasynaptic NMDA receptors (NMDARs) which mediate Na+ and Ca2+ influx into neurons and regulate membrane polarization [82].
2.3. Tau Protein in the Nucleus of Neurons
Tau is localized in the nucleus of neurons, more precisely, in nucleolar organized regions of the nucleolus [83][84][85][86]. Since nucleolus is the center of ribosomal RNA (rRNA) synthesis and processing, several studies which described localization of tau in parts of nucleolus suggest that tau can be involved in rRNA-coding DNA transcription and rRNA processing [86][87]. Tau can directly bind DNA [88] and RNA [89] and protects them from oxidative damage [90][91] which helps to maintain DNA and RNA integrity [92]. Furthermore, tau-DNA interaction is modulated by tau phosphorylation, which strongly reduces the ability of tau to bind DNA [93]. Hyperphosphorylation, which is one of the major hallmarks of the pathological forms of tau, may also influence its nucleocytoplasmic transport. According to the recent study, hyperphosphorylated tau directly interacts with a subunit of nuclear pore complex—nucleoporin NUP98 causes its mislocalization and disrupts the nucleocytoplasmic transport [94].
2.4. Big Tau
Big tau was initially known from studies on rats and mice where tau proteins were detected in their tissues, as well as, in cell lines derived from these species. This high-molecular-weight form of a rat and murine tau has an apparent molecular weight of ~110 kDa and possesses 733 or 752 amino acids (aa), respectively [95][96], as a result of the involvement of exon 4a and alternatively spliced exon 6 of MAPT gene in its transcript [97][98]. It was shown that Big tau is expressed only in the peripheral nervous system (PNS), neurons of the optic nerve, but also in specific CNS neurons with long axons projecting to the periphery [96][99]. In human sequence databases, Big tau is also designated as the PNS tau. Human Big tau (Figure 3) has not been confirmed experimentally in human tissues or human-derived cell lines so far, and its specific function remains unanswered. In light of the known functions of brain tau protein, several benefits of Big tau due to its increased length were proposed. The most important is the increased spacing between microtubules observed in processes of Sf9 cells overexpressing Big tau, which may reduce the energy required for axonal transport [100]. Furthermore, the elongated N-terminus of Big tau was proposed to reduce the rate of phosphorylation of motor proteins, and not mitigating their activity, thus supporting uninterrupted axonal transport [97][101].
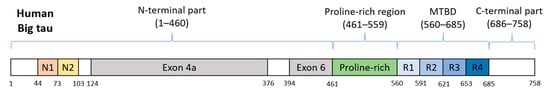
Figure 3. Schematic representation of human Big tau. Its counterparts in rat and mouse possess 752 or 733 aa and share 74.9% or 74.2% identity, respectively. Exons 4a and 6 are responsible for differentiation from the brain tau protein.
2.5. Extracellular Tau Protein
Mounting evidence shows that pathological forms of tau protein spread from diseased to healthy cells and transform physiological tau to its misfolded pathological forms which in turn self-aggregate and form PHF and NFT [102][103]. It was shown that neuronal activity, accompanied by synaptic transmission, mediates tau protein release into extracellular space, mainly in an exosome-bound form, but also in a soluble form, and that this process occurs under both physiological and pathological conditions [63][64][65][66]. One of the suggested pathways of pathological tau spreading is connected to the resident macrophages of CNS–microglia [73]. Microglia have both phagocytic and secretion properties and can play a key role in the spreading of tau pathology [104]. Microglial exosomes serve as a medium for intercellular transport of cytokines, miRNAs, and other regulating factors [105]. The fact that microglia can be involved in the spreading of pathological tau forms is supported by several experiments. The group of Asai showed that depletion of the microglia in mouse brain significantly slowed propagation of tau between cells and that tau spreading was mediated by microglial exosomes [106]. Another research group demonstrated that reactive, inflammatory microglia can contribute to the spreading of tau pathology [73].
References
- Uversky, V.N. Intrinsic disorder, protein–protein interactions, and disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121.
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862.
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247.
- Ferreira, A.; Busciglio, J.; Cáceres, A. Microtubule formation and neurite growth in cerebellar macroneurons which develop in vitro: Evidence for the involvement of the microtubule-associated proteins, MAP-1a, HMW-MAP2 and Tau. Dev. Brain Res. 1989, 49, 215–228.
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000, 150, 989–1000.
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91.
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21.
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378.
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373.
- Müller, R.; Heinrich, M.; Heck, S.; Blohm, D.; Richter-Landsberg, C. Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 1997, 288, 239–249.
- Komori, T. Tau-positive dial Inclusions in Progressive Supranuclear Palsy, Corticobasal Degeneration and Pick’s Disease. Brain Pathol. 1999, 9, 663–679.
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689.
- Trojanowski, J.Q.; Schuck, T.; Schmidt, M.L.; Lee, V. Distribution of tau proteins in the normal human central and peripheral nervous system. J. Histochem. Cytochem. 1989, 37, 209–215.
- Dugger, B.N.; Hoffman, B.R.; Scroggins, A.; Serrano, G.E.; Adler, C.H.; Shill, H.A.; Belden, C.M.; Sabbagh, M.N.; Caviness, J.N.; Dunckley, E.D. Tau immunoreactivity in peripheral tissues of human aging and select tauopathies. Neurosci. Lett. 2019, 696, 132–139.
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230.
- Kosik, K.S.; Orecchio, L.D.; Bakalis, S.; Neve, R.L. Developmentally regulated expression of specific tau sequences. Neuron 1989, 2, 1389–1397.
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130.
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704.
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288.
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506.
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114 Pt 6, 1179–1187.
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491.
- Tan, D.C.S.; Yao, S.; Ittner, A.; Bertz, J.; Ke, Y.D.; Ittner, L.M.; Delerue, F. Generation of a New Tau Knockout (tauDeltaex1) Line Using CRISPR/Cas9 Genome Editing in Mice. J. Alzheimer’s Dis. 2018, 62, 571–578.
- Terwel, D.; Lasrado, R.; Snauwaert, J.; Vandeweert, E.; Van Haesendonck, C.; Borghgraef, P.; Van Leuven, F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J. Biol. Chem. 2005, 280, 3963–3973.
- Tucker, K.L.; Meyer, M.; Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37.
- DiTella, M.; Feiguin, F.; Carri, N.; Kosik, K.; Caceres, A. MAP-1B/TAU functional redundancy during laminin-enhanced axonal growth. J. Cell Sci. 1996, 109, 467–477.
- Lei, P.; Ayton, S.; Moon, S.; Zhang, Q.; Volitakis, I.; Finkelstein, D.I.; Bush, A.I. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 2014, 9, 29.
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478.
- Hong, X.P.; Peng, C.X.; Wei, W.; Tian, Q.; Liu, Y.H.; Yao, X.Q.; Zhang, Y.; Cao, F.Y.; Wang, Q.; Wang, J.Z. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 2010, 20, 1339–1349.
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295.
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130144.
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812.
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397.
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Heraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940.
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711.
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754.
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Muller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473.
- De Vos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 2013, 33, 12887–12897.
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014, 76, 443–456.
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278.
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259.
- Lin, W.-L.; Lewis, J.; Yen, S.-H.; Hutton, M.; Dickson, D.W. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am. J. Pathol. 2003, 162, 213–218.
- Perea, J.R.; López, E.; Díez-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci. 2019, 13, 442.
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2018, 145, 355–368.
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204.
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17.
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114.
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; La Ferla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853.
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472.
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018, 22, 832–847.
- Rademakers, R.; Cruts, M.; van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 2004, 24, 277–295.
- Tolnay, M.; Probst, A. Argyrophilic grain disease. Handb. Clin. Neurol. 2008, 89, 553–563.
- Mahapatra, R.K.; Edwards, M.J.; Schott, J.M.; Bhatia, K.P. Corticobasal degeneration. Lancet Neurol. 2004, 3, 736–743.
- Arai, T.; Ikeda, K.; Akiyama, H.; Nonaka, T.; Hasegawa, M.; Ishiguro, K.; Iritani, S.; Tsuchiya, K.; Iseki, E.; Yagishita, S.; et al. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann. Neurol. 2004, 55, 72–79.
- Hauw, J.J.; Verny, M.; Delaere, P.; Cervera, P.; He, Y.; Duyckaerts, C. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer’s disease and aging. Neurosci. Lett. 1990, 119, 182–186.
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340.
- Brandt, R.; Trushina, N.I.; Bakota, L. Much More Than a Cytoskeletal Protein: Physiological and Pathological Functions of the Non-microtubule Binding Region of Tau. Front. Neurol. 2020, 11, 1–14.
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089.
- Konzack, S.; Thies, E.; Marx, A.; Mandelkow, E.M.; Mandelkow, E. Swimming against the tide: Mobility of the microtubule-associated protein tau in neurons. J. Neurosci. 2007, 27, 9916–9927.
- Utton, M.A.; Noble, W.J.; Hill, J.E.; Anderton, B.H.; Hanger, D.P. Molecular motors implicated in the axonal transport of tau and α-synuclein. J. Cell Sci. 2005, 118, 4645–4654.
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554.
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Takashima, A. Enhanced tau protein translation by hyper-excitation. Front. Aging Neurosci. 2019, 11, 322.
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393.
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 1–25.
- Sato, C.; Barthelemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e7.
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394.
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215.
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431.
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63.
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316.
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527.
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980.
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755.
- Mondragon-Rodriguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053.
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435.
- Suetsugu, S.; Toyooka, K.; Senju, Y. Subcellular Membrane Curvature Mediated by the BAR Domain Superfamily Proteins. Semin. Cell Dev. Biol. 2010, 21, 340–349.
- Sinsky, J.; Majerova, P.; Kovac, A.; Kotlyar, M.; Jurisica, I.; Hanes, J. Physiological tau interactome in brain and its link to tauopathies. J. Proteome Res. 2020, 19, 2429–2442.
- Safari, F.; Suetsugu, S. The BAR domain superfamily proteins from subcellular structures to human diseases. Membranes 2012, 2, 91–117.
- Liu, Y.; Lv, K.; Li, Z.; Yu, A.C.; Chen, J.; Teng, J. PACSIN1, a Tau-interacting protein, regulates axonal elongation and branching by facilitating microtubule instability. J. Biol. Chem. 2012, 287, 39911–39924.
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009, 10, 1–12.
- Elie, A.; Prezel, E.; Guerin, C.; Denarier, E.; Ramirez-Rios, S.; Serre, L.; Andrieux, A.; Fourest-Lieuvin, A.; Blanchoin, L.; Arnal, I. Tau co-organizes dynamic microtubule and actin networks. Sci. Rep. 2015, 5, 9964.
- Pallas-Bazarra, N.; Draffin, J.; Cuadros, R.; Antonio Esteban, J.; Avila, J. Tau is required for the function of extrasynaptic NMDA receptors. Sci. Rep. 2019, 9, 9116.
- Lu, J.; Li, T.; He, R.; Bartlett, P.F.; Gotz, J. Visualizing the microtubule-associated protein tau in the nucleus. Sci. China Life Sci. 2014, 57, 422–431.
- Liu, C.; Gotz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849.
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 2018, 6, 70.
- Sjoberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119 Pt 10, 2025–2034.
- Thurston, V.C.; Zinkowski, R.P.; Binder, L.I. Tau as a nucleolar protein in human nonneural cells in vitro and in vivo. Chromosoma 1996, 105, 20–30.
- Wei, Y.; Qu, M.H.; Wang, X.S.; Chen, L.; Wang, D.L.; Liu, Y.; Hua, Q.; He, R.Q. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE 2008, 3, e2600.
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349.
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84.
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575.
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Begard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047.
- Qi, H.; Cantrelle, F.X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buee, L.; Lippens, G.; Bonnefoy, E.; Galas, M.C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533.
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 2018, 99, 925–940.e7.
- Goedert, M.; Spillantini, M.; Crowther, R. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987.
- Taleghany, N.; Oblinger, M. Regional distribution and biochemical characteristics of high molecular weight tau in the nervous system. J. Neurosci. Res. 1992, 33, 257–265.
- Fischer, I.; Baas, P.W. Resurrecting the mysteries of big tau. Trends Neurosci. 2020, 43, 493–504.
- Mercken, M.; Fischer, I.; Kosik, K.; Nixon, R. Three distinct axonal transport rates for tau, tubulin, and other microtubule-associated proteins: Evidence for dynamic interactions of tau with microtubules in vivo. J. Neurosci. 1995, 15, 8259–8267.
- Boyne, L.; Tessler, A.; Murray, M.; Fischer, I. Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293.
- Frappier, T.F.; Georgieff, I.S.; Brown, K.; Shelanski, M.L. τ Regulation of Microtubule-Microtubule Spacing and Bundling. J. Neurochem. 1994, 63, 2288–2294.
- Kanaan, N.M.; Morfini, G.; Pigino, G.; LaPointe, N.E.; Andreadis, A.; Song, Y.; Leitman, E.; Binder, L.I.; Brady, S.T. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 2012, 33, 826.e15–826.e30.
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852.
- Clavaguera, F.; Duyckaerts, C.; Haïk, S. Prion-like properties of Tau assemblies. Curr. Opin. Neurobiol. 2020, 61, 49–57.
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118.
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell communication by extracellular vesicles: Focus on microglia. Neuroscience 2019, 405, 148–157.
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.3K
Revisions:
2 times
(View History)
Update Date:
15 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No