Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hannah Crossland | + 2345 word(s) | 2345 | 2021-09-13 11:08:19 | | | |
| 2 | Camila Xu | Meta information modification | 2345 | 2021-12-13 04:31:59 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Crossland, H. The Regulatory Roles of PPARs. Encyclopedia. Available online: https://encyclopedia.pub/entry/16989 (accessed on 28 July 2026).
Crossland H. The Regulatory Roles of PPARs. Encyclopedia. Available at: https://encyclopedia.pub/entry/16989. Accessed July 28, 2026.
Crossland, Hannah. "The Regulatory Roles of PPARs" Encyclopedia, https://encyclopedia.pub/entry/16989 (accessed July 28, 2026).
Crossland, H. (2021, December 10). The Regulatory Roles of PPARs. In Encyclopedia. https://encyclopedia.pub/entry/16989
Crossland, Hannah. "The Regulatory Roles of PPARs." Encyclopedia. Web. 10 December, 2021.
Copy Citation
Peroxisome proliferator-activated receptors (PPARs) are a group of transcription factors implicated in wide-ranging cellular functions, including lipid metabolism, inflammatory responses and cell proliferation and differentiation.
skeletal muscle
inflammation
PPARs
substrate metabolism
1. Introduction
Peroxisome proliferator-activated receptors (PPARs) are a group of transcription factors implicated in wide-ranging cellular functions, including lipid metabolism, inflammatory responses and cell proliferation and differentiation [1]. Three PPAR subtypes exist (PPARα, PPARδ and PPARγ). They are activated in vivo by endogenous fatty acids and their metabolites and synthetic compounds developed for their lipid-lowering and anti-diabetic actions. Skeletal muscle is a tissue that displays high metabolic flexibility, comprising different fibre types that vary according to their contractile and metabolic properties [2]. For example, slow-twitch type I fibres have a relatively high capillary density, are rich in mitochondria and possess a relatively high capacity for oxidative metabolism during contraction. In contrast, fast-twitch type IIx fibres have a lower capillary density and a high capacity for energy delivery from non-mitochondrial routes during contraction. Disturbances in skeletal muscle energy homeostasis play a key part in the pathogenesis of several chronic non-communicable disease conditions, including type 2 diabetes (T2D) and chronic lung disease. The dysregulation of skeletal muscle energy homeostasis is also a frequent underlying characteristic of acute inflammation-related conditions, such as sepsis [3] and surgical trauma. The role of PPAR agonism in modulating skeletal muscle protein and fuel metabolism in these conditions is relatively poorly understood.
2. Metabolic Functions of PPARs and Their Actions in Skeletal Muscle
Peroxisome proliferator-activated receptors (PPARs) are a group of proteins that belong to the nuclear hormone receptor superfamily of ligand-activated transcription factors. PPAR transcriptional activity is mediated by heterodimers of PPARs with retinoid X receptor (RXR), which subsequently bind to DNA sequence elements (PPREs) in regulatory regions of target genes [4]. Three PPAR subtypes have been identified (PPARα, PPARδ (also known as PPARβ) and PPARγ [5]. Through their interactions with endogenous lipids and lipid metabolites, PPARs have been reported to regulate many metabolic processes, including lipid and glucose homeostasis, cell proliferation and inflammation [1]. Several endogenous compounds, including n-3 and n-6 fatty acids, eicosanoids and phospholipids, have been identified as natural ligands of PPARs. In addition to this, activation of PPAR activity by pharmacological agonists has been identified as a promising treatment strategy for conditions related to insulin resistance and dyslipidaemia, in part through increased fatty acid oxidation in skeletal muscle, thereby decreasing overall body fat content [6].
Each PPAR subtype has been attributed to different tissue-specific expression levels and functions. For example, PPARα is highly expressed in tissue types that undergo significant fatty acid catabolism, such as brown adipose tissue, heart and liver [7]. Activated by polyunsaturated fatty acids (PUFA) and leukotriene, PPARα has an important function in fatty acid catabolism and carbohydrate metabolism [8][9]. Synthetic compounds that act as agonists of PPARα are known as fibrates, whose actions are important in lipid-lowering activities and cardio-protection [10][11]. PPARγ, on the other hand, is variably expressed in adipocytes, macrophages, placenta and other tissues and is activated by specific endogenous fatty acid metabolites (such as 15-deoxy-prostaglandin J2) as well as by a class of insulin sensitisers known as thiazolidinediones (TZDs) [12]. TZDs have been proven to be important in the treatment of T2D. While early TZDs (e.g., Troglitazone) were related to severe hepatic side effects, other newer available TZDs (Rosiglitazone, Pioglitazone) are not toxic to the liver. PPARγ plays a central role in adipogenesis, whereby the insulin-sensitising effect of TZDs may be due to new adipose cell recruitment, enabling increased lipid storage capacity and adipokine secretion [13]. PPARγ activation also regulates the transcription of genes that promote the synthesis of triglycerides [13]. In patients with T2D, administration of TZDs successfully improved insulin-stimulated glucose disposal under euglycaemic-hyperinsulinaemic clamp conditions [14][15], where skeletal muscle plays a central glucose-lowering role. One mechanism by which TZDs exert their insulin sensitising actions on skeletal muscle is through the modulation of adipose secretory factors, such as adiponectin. Increased secretion of adiponectin has been suggested to act as an insulin sensitiser for liver and skeletal muscle, and this occurs through the activation of PPARγ [16].
The role of PPARδ has remained relatively unclear until recently, where it has been associated with a wide range of metabolic functions in vivo [17][18]. It has broad expression across tissues and is activated by various ligands, such as long-chain fatty acids. A developmental regulatory role has been identified for PPARδ, as well as regulation of lipid metabolism [17][18]. It is the predominant isotype in skeletal muscle, where it has been linked to fuel metabolism, energy expenditure, inflammation, and fibre type switching through physical exercise [17][19]. Both PPARα and PPARδ have been demonstrated to regulate genes for proteins involved in fatty acid uptake and oxidation, including lipoprotein lipase (LPL), fatty acid-binding protein 3 (FABP), stearoyl-Coenzyme A desaturase (SCD)-1 and cluster of differentiation 36 (CD36) [20][21]. During fasting, PPARδ expression is upregulated in rodent skeletal muscles, which is important in regulating the cellular uptake and oxidation of free fatty acids (FFA) as an energy source for ATP production [22].
Through their importance in metabolic regulation, the role of all three PPAR subtypes in skeletal muscle metabolism has been established. For example, one link between PPARs and metabolic regulation in skeletal muscle appears to be through the upregulation of pyruvate dehydrogenase kinase 4 (PDK4), a key regulator of the pyruvate dehydrogenase complex (PDC). The PDC activation status is regulated by various competing PDKs and pyruvate dehydrogenase phosphatase (PDP) proteins [23]. These covalent processes ultimately determine the extent of PDC phosphorylation (i.e., activation). There are four isoforms of PDK (PDK1-4) and two isoforms of PDP (PDP1 and 2) [24][25]. While PDK1 and PDK3 appear to be mainly expressed in the heart, pancreatic islet cells and kidney, PDK2 and PDK4 are expressed in most tissues, including heart and skeletal muscle [24]. Selective PDK4 upregulation has been demonstrated in response to starvation conditions and pathologies such as T2D [26][27], which is thought to be due to changes in FFA availability in skeletal muscle. An increase in fatty acid oxidation via PPARδ agonism [6], and starvation [28], is believed to be responsible for the PDK4 transcriptional activation, thereby inactivating PDC (the rate-limiting enzyme in mitochondrial carbohydrate oxidation). It should be noted, however, that a lack of association between increases in plasma FFA levels and muscle PDK4 expression has been reported during fasting in humans [29], with no observable changes in muscle PPARα expression, indicating that other factors could also be important in PDK4 upregulation. One such factor could be the Forkhead box class O (FOXO) family of transcription factors, which has been linked to promoter binding of the PDK4 gene as a result of FFA-mediated nuclear translocation [30].
In addition to increased availability of endogenous fatty acids and their metabolites being associated with PPAR activation, inflammation has been proven to be a major site of PPAR regulation, which can occur through both direct and indirect mechanisms [31]. As mentioned, PPARs have emerged as targets of drugs used to treat various aspects of the metabolic syndrome, of which inflammation is an underlying key factor. All three PPAR isotypes have been shown to exert anti-inflammatory effects during conditions of chronic low-grade inflammation, characterised by increased circulatory cytokines and acute-phase proteins [32][33]. PPARα was shown to upregulate the expression of IkB, a factor that suppresses the nuclear translocation and transcriptional activity of the pro-inflammatory nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) [34]. PPARγ has also been shown to reduce activation of NF-kB, as well as inhibit pro-inflammatory cytokine production in T lymphocytes and induction of anti-inflammatory regulatory molecules of the innate immune system [35].
In summary, all three PPAR subtypes have distinct yet overlapping roles in regulating metabolic function and inflammation (see Table 1), and synthetic compounds aimed at activating the PPARs have been developed for their lipid-lowering and anti-diabetic actions. In skeletal muscle, PPAR activation appears important in the upregulation of PDK4, thereby demonstrating its essential role in regulating carbohydrate oxidation and energy homeostasis. The following section of this review will focus in more detail on the impact of PPARδ agonism on muscle metabolism and contractile function and PPARγ agonism on muscle metabolism and inflammation.
Table 1. Regulation of lipid and carbohydrate metabolism by PPARs in skeletal muscle, adipose tissue and liver.
| Skeletal Muscle | Liver | Adipose | |
|---|---|---|---|
| PPARδ | + FA oxidation −carbohydrate oxidation |
+ FA oxidation − lipogenesis |
+ FA oxidation |
| PPARγ | + FA oxidation + glucose uptake |
+ lipogenesis + lipid storage − glucose production |
+ adipogenesis + lipogenesis + lipid storage + adipokine production |
| PPARα | + FA oxidation | + FA oxidation − lipid storage |
3. PPARδ Agonism and Skeletal Muscle Metabolism, Contractile Function and Inflammation
Several in vivo animal studies have been performed with the aim of determining the impact of PPARδ agonism on skeletal muscle metabolism and function. We previously demonstrated in our laboratory that 6 days of administration of the PPARδ agonist, GW610742 [36], resulted in increased activity of β-hydroxy acyl-CoA dehydrogenase (β-HAD) in resting rat soleus muscle, which is a key step in β-oxidation in the mitochondria. Compared with control animals, these changes were paralleled by increased expression of muscle PDK2 and PDK4 mRNA and PDK4 protein expression. Thus, evidence points towards PPAR activation in skeletal muscle being, in part, important in mediating FFA-induced PDK4 upregulation in skeletal muscle, thereby contributing to PDC inhibition, suppressing PDC-regulated carbohydrate oxidation, and switching fuel selection towards fat oxidation in skeletal muscle (Figure 1). We also measured the impact of GW610742 on muscle growth-related pathways since FOXO1, which plays a part in PDK4 upregulation, has also been suggested to increase transcription of MAFbx and MuRF1, thereby activating ubiquitin-proteasome mediated muscle proteolysis [37]. In keeping with this, administration of the PPARδ agonist resulted in increases in muscle mRNA and protein expression of MAFbx and MuRF1, suggesting that potentially the induction of muscle atrophy signalling is another consequence of PPARδ agonism. Collectively, the findings pointed to PPARδ agonism being involved in the regulation of muscle fuel selection and the induction of a muscle atrophy programme via a single common signalling pathway. It should be stated, however, there was no evidence of soleus muscle atrophy based on the muscle protein:DNA ratio after 6 days of GW610742 administration compared with control.
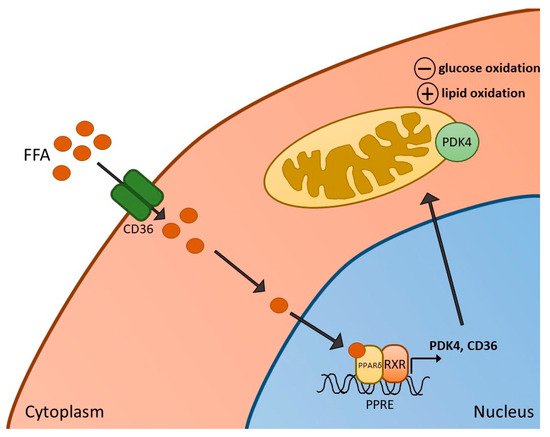
Figure 1. Activation of PPARδ in skeletal muscle. Increased free fatty acids (FFA) and their metabolites enter skeletal muscle via the FFA transporter CD36, resulting in the formation of a heterodimer of PPARδ and retinoid X receptor (RXR), and subsequent activation of PPARδ-dependent genes, such as pyruvate dehydrogenase kinase 4 (PDK4) (and CD36 itself). Activation of PDK4 can result in reduced rates of glucose oxidation as well as increased fatty acid oxidation in mitochondria.
In line with the above findings relating to a PPARδ agonism induced switch in muscle fuel selection away from carbohydrate to increased fat oxidation, in another study, mice treated with the PPARδ agonist GW501516 exhibited increased PGC-1α levels, and improved prolonged low-intensity wheel-running performance. They also saw hypertrophy of oxidative slow-twitch myofibres, which are rich in mitochondria, perhaps suggesting increased reliance on the catabolism of FA through mitochondrial beta-oxidation [38]. However, we further reported that when muscle contraction was increased to an intensity that necessitates carbohydrate to become an obligate fuel for contraction, PPARδ agonism negatively affected contractile function in rats [39]. Specifically, male Wistar rats received the PPARδ agonist GW610742X (or vehicle) for 6 days. The gastrocnemius–soleus–plantaris muscle group was isolated and subjected to submaximal electrically evoked contraction using a perfused hindlimb model. The contraction intensity was fixed to guarantee carbohydrate become an essential fuel, and PDC activity was increased, ensuring pyruvate derived acetyl group delivery to the mitochondrion [40]. We observed that PDC activity during contraction was significantly less with the PPARδ agonist than control, while anaerobic metabolism (reflected by phosphocreatine hydrolysis and lactate accumulation) was greater. We proposed that this collectively accounted for the observed impaired contractile function with GW610742X agonist, indicating that PPARδ agonism can impair the contractile muscle function by inhibiting carbohydrate oxidation during muscle contraction where carbohydrate is an obligate fuel.
We have also sought to determine whether PPAR transcription factors may be necessary for the high-fat feeding induced inhibition of PDC activation and carbohydrate oxidation during submaximal exercise in humans [41][42]. Healthy male volunteers were given a control diet or an iso-caloric high-fat diet (HFD). They underwent 60 min of submaximal exercise at an intensity equivalent to 75% maximal oxygen uptake. There was a relative increase in expression of PDK4 in muscle with HFD compared to control, alongside reduced PDC activation in muscle. Exercise increased PDC activity and carbohydrate utilisation with both diets, but these measures were diminished with the HFD. In terms of PPAR expression, there was no effect of the high-fat diet on the mRNA expression of PPARδ. However, PPARγ and PPARα were increased at rest, though this increase was not apparent during exercise. Of note, the expression of PPARα mRNA was lower in another group of volunteers that underwent a HFD but were also treated with dichloroacetate (DCA), a potent inhibitor of PDK2 and PDK4 and, therefore, a stimulator of PDC activity, which restored carbohydrate oxidation during exercise in this group. Thus, in humans, these results appear to suggest there may not be significant involvement of PPARs in increasing muscle PDK4 expression, although muscle protein levels of each PPAR were not measured.
Following these findings, further work from our laboratory studied the role of PPARδ (and FOXO1) in palmitate-induced PDC inhibition and carbohydrate use using a skeletal muscle cell model [43]. Myotubes were treated with palmitate for 16 hrs in the presence or absence of continuous electrical pulse stimulation, the latter having been shown to increase glucose uptake and carbohydrate oxidation in muscle cells [44][45], and therefore potentially having the capacity to reverse palmitate-mediated inhibition of PDC. It was observed that palmitate reduced glucose uptake, PDC activity and maximal rates of palmitate derived mitochondrial ATP production whilst also increasing PDK4, PPARδ and PPARα proteins. There was also a significant reduction in the magnitude of FOXO1 phosphorylation, indicating its nuclear translocation and subsequent activation. Electrical pulse stimulation reversed many palmitate-induced changes to carbohydrate oxidation and was associated with reduced PDK4 protein and reduced PPARδ (but not PPARα) protein content. Collectively, while more work is required to elucidate the relative importance of PPARδ and FOXO1 transcription factors in mediating PDK4 transcription, particularly in humans, these findings indicate their potential roles in palmitate-induced impairments in PDC activity and carbohydrate oxidation in skeletal muscle.
References
- Berger, J.; Wagner, J.A. Physiological and Therapeutic Roles of Peroxisome Proliferator-Activated Receptors. Diabetes Technol. Ther. 2002, 4, 163–174.
- Mukund, K.; Subramaniam, S. Skeletal muscle: A review of molecular structure and function, in health and disease. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1462.
- Puthucheary, Z.A.; Rawal, J.; McPhail, M.; Connolly, B.; Ratnayake, G.; Chan, P.; Hopkinson, N.S.; Padhke, R.; Dew, T.; Sidhu, P.S.; et al. Acute Skeletal Muscle Wasting in Critical Illness. JAMA 2013, 310, 1591–1600.
- Varga, T.; Czimmerer, Z.; Nagy, L. PPARs are a unique set of fatty acid regulated transcription factors controlling both lipid metabolism and inflammation. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2011, 1812, 1007–1022.
- Lee, C.-H.; Olson, P.; Evans, R.M. Minireview: Lipid Metabolism, Metabolic Diseases, and Peroxisome Proliferator-Activated Receptors. Endocrinology 2003, 144, 2201–2207.
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Watanabe, M.; Magoori, K.; Ioka, R.X.; Tachibana, K.; et al. Activation of peroxisome proliferator-activated receptor δ induces fatty acid β-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929.
- Abbott, B.D. Review of the expression of peroxisome proliferator-activated receptors alpha (PPARα), beta (PPARβ), and gamma (PPARγ) in rodent and human development. Reprod. Toxicol. 2009, 27, 246–257.
- Baes, M.; Peeters, A. Role of PPARα in hepatic carbohydrate metabolism. PPAR Res. 2010, 2010, 572405.
- Muoio, D.M.; MacLean, P.S.; Lang, D.B.; Li, S.; Houmard, J.A.; Way, J.M.; Winegar, D.A.; Corton, J.C.; Dohm, G.L.; Kraus, W.E. Fatty Acid Homeostasis and Induction of Lipid Regulatory Genes in Skeletal Muscles of Peroxisome Proliferator-activated Receptor (PPAR) α Knock-out Mice. Evidence for compensatory regulation by PPARδ. J. Biol. Chem. 2002, 277, 26089–26097.
- Ueno, H.; Saitoh, Y.; Mizuta, M.; Shiiya, T.; Noma, K.; Mashiba, S.; Kojima, S.; Nakazato, M. Fenofibrate ameliorates insulin resistance, hypertension and novel oxidative stress markers in patients with metabolic syndrome. Obes. Res. Clin. Pr. 2011, 5, e335–e340.
- Koh, K.K.; Han, S.H.; Quon, M.J.; Ahn, J.Y.; Shin, E.K. Beneficial Effects of Fenofibrate to Improve Endothelial Dysfunction and Raise Adiponectin Levels in Patients With Primary Hypertriglyceridemia. Diabetes Care 2005, 28, 1419–1424.
- Fajas, L.; Auboeuf, D.; Raspé, E.; Schoonjans, K.; Lefebvre, A.-M.; Saladin, R.; Najib, J.; Laville, M.; Fruchart, J.-C.; Deeb, S.; et al. The Organization, Promoter Analysis, and Expression of the Human PPARγ Gene. J. Biol. Chem. 1997, 272, 18779–18789.
- El Akoum, S. PPAR Gamma at the Crossroads of Health and Disease: A Masterchef in Metabolic Homeostasis. Endocrinol. Metab. Syndr. 2014, 3, 2161–1017.
- Miyazaki, Y.; He, H.; Mandarino, L.J.; DeFronzo, R.A. Rosiglitazone improves downstream insulin receptor signaling in type 2 diabetic patients. Diabetes 2003, 52, 1943–1950.
- Gastaldelli, A.; Ferrannini, E.; Miyazaki, Y.; Matsuda, M.; Mari, A.; DeFronzo, R.A. Thiazolidinediones improve β-cell function in type 2 diabetic patients. Am. J. Physiol. Metab. 2007, 292, E871–E883.
- Yamauchi, T.; Kamon, J.; Waki, H.; Terauchi, Y.; Kubota, N.; Hara, K.; Mori, Y.; Ide, T.; Murakami, K.; Tsuboyama-Kasaoka, N.; et al. The fat-derived hormone adiponectin reverses insulin re-sistance associated with both lipoatrophy and obesity. Nat. Med. 2001, 7, 941–946.
- Ehrenborg, E.; Krook, A. Regulation of Skeletal Muscle Physiology and Metabolism by Peroxisome Proliferator-Activated Receptor δ. Pharmacol. Rev. 2009, 61, 373–393.
- Tan, N.S.; Vázquez-Carrera, M.; Montagner, A.; Sng, M.K.; Guillou, H.; Wahli, W. Transcriptional control of physiological and pathological processes by the nuclear receptor PPARβ/δ. Prog. Lipid Res. 2016, 64, 98–122.
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An apparent functional consequence in skeletal muscle physiology via pe-roxisome proliferator-activated receptors. Int. J. Mol. Sci. 2018, 19, 1425.
- Pawlak, M.; Lefebvre, P.; Staels, B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2015, 62, 720–733.
- Dressel, U.; Allen, T.L.; Pippal, J.B.; Rohde, P.R.; Lau, P.; Muscat, G.E.O. The Peroxisome Proliferator-Activated Receptor β/δ Agonist, GW501516, Regulates the Expression of Genes Involved in Lipid Catabolism and Energy Uncoupling in Skeletal Muscle Cells. Mol. Endocrinol. 2003, 17, 2477–2493.
- Peters, S.J.; Harris, R.A.; Heigenhauser, G.J.F.; Spriet, L.L. Muscle fiber type comparison of PDH kinase activity and isoform expression in fed and fasted rats. Am. J. Physiol. Integr. Comp. Physiol. 2001, 280, R661–R668.
- Wieland, O.H. The mammalian pyruvate dehydrogenase complex: Structure and regulation. Rev. Physiol. Biochem. Pharmacol. 1983, 96, 123–170.
- Bowker-Kinley, M.M.; Davis, I.W.; Wu, P.; Harris, A.R.; Popov, M.K. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998, 329, 191–196.
- Huang, B.; Gudi, R.; Wu, P.; Harris, R.A.; Hamilton, J.; Popov, K.M. Isoenzymes of Pyruvate Dehydrogenase Phosphatase. DNA-derived amino acid sequences, expression, and regulation. J. Biol. Chem. 1998, 273, 17680–17688.
- Sugden, M.C.; Kraus, A.; Harris, R.A.; Holness, M.J. Fibre-type specific modification of the activity and regulation of skeletal muscle pyruvate dehydrogenase kinase (PDK) by prolonged starvation and refeeding is associated with targeted regulation of PDK isoenzyme 4 expression. Biochem. J. 2000, 346, 651–657.
- Wu, P.; Inskeep, K.; Bowker-Kinley, M.M.; Popov, K.M.; Harris, R.A. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999, 48, 1593–1599.
- Tsintzas, K.; Jewell, K.; Kamran, M.; Laithwaite, D.; Boonsong, T.; Littlewood, J.; Macdonald, I.; Bennett, A. Differential regulation of metabolic genes in skeletal muscle during starvation and refeeding in humans. J. Physiol. 2006, 575, 291–303.
- Spriet, L.L.; Tunstall, R.J.; Watt, M.J.; Mehan, K.A.; Hargreaves, M.; Cameron-Smith, D. Pyruvate dehydrogenase activation and kinase expression in human skeletal muscle during fasting. J. Appl. Physiol. 2004, 96, 2082–2087.
- Furuyama, T.; Kitayama, K.; Yamashita, H.; Mori, N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem. J. 2003, 375, 365–371.
- Wahli, W.; Michalik, L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol. Metab. 2012, 23, 351–363.
- Michalik, L.; Wahli, W. PPARs Mediate Lipid Signaling in Inflammation and Cancer. PPAR Res. 2008, 2008, 134059.
- Bishop-Bailey, D.; Bystrom, J. Emerging roles of peroxisome proliferator-activated receptor-β/δ in inflammation. Pharmacol. Ther. 2009, 124, 141–150.
- Delerive, P.; De Bosscher, K.; Berghe, W.V.; Fruchart, J.-C.; Haegeman, G.; Staels, B. DNA Binding-Independent Induction of IκBα Gene Transcription by PPARα. Mol. Endocrinol. 2002, 16, 1029–1039.
- Huang, W.; Glass, C.K. Nuclear receptors and inflammation control: Molecular mechanisms and pathophysiological rele-vance. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1542–1549.
- Constantin, D.; Constantin-Teodosiu, D.; Layfield, R.; Tsintzas, K.; Bennett, A.J.; Greenhaff, P.L. PPARδ agonism induces a change in fuel metabolism and activation of an atrophy programme, but does not impair mitochondrial function in rat skeletal muscle. J. Physiol. 2007, 583, 381–390.
- Léger, B.; Cartoni, R.; Praz, M.; Lamon, S.; Dériaz, O.; Crettenand, A.; Gobelet, C.; Rohmer, P.; Konzelmann, M.; Luthi, F.; et al. Akt signalling through GSK-3β, mTOR and Foxo1 is involved in human skeletal muscle hypertrophy and atrophy. J. Physiol. 2006, 576, 923–933.
- Chen, W.; Gao, R.; Xie, X.; Zheng, Z.; Li, H.; Li, S.; Dong, F.; Wang, L. A metabolomic study of the PPARδ agonist GW501516 for enhancing running endurance in Kunming mice. Sci. Rep. 2015, 5, 9884.
- Constantin-Teodosiu, D.; Baker, D.J.; Constantin, D.; Greenhaff, P.L. PPARδ agonism inhibits skeletal muscle PDC activity, mitochondrial ATP production and force generation during prolonged contraction. J. Physiol. 2009, 587, 231–239.
- Constantin-Teodosiu, D.; Cederblad, G.; Hultman, E. PDC activity and acetyl group accumulation in skeletal muscle during isometric contraction. J. Appl. Physiol. 1993, 74, 1712–1718.
- Constantin-Teodosiu, D.; Constantin, D.; Stephens, F.; Laithwaite, D.; Greenhaff, P.L. The Role of FOXO and PPAR Transcription Factors in Diet-Mediated Inhibition of PDC Activation and Carbohydrate Oxidation During Exercise in Humans and the Role of Pharmacological Activation of PDC in Overriding These Changes. Diabetes 2012, 61, 1017–1024.
- Constantin-Teodosiu, D. Regulation of Muscle Pyruvate Dehydrogenase Complex in Insulin Resistance: Effects of Exercise and Dichloroacetate. Diabetes Metab. J. 2013, 37, 301–314.
- Chien, H.-C.; Greenhaff, P.L.; Constantin-Teodosiu, D. PPARδ and FOXO1 Mediate Palmitate-Induced Inhibition of Muscle Pyruvate Dehydrogenase Complex and CHO Oxidation, Events Reversed by Electrical Pulse Stimulation. Int. J. Mol. Sci. 2020, 21, 5942.
- Grosset, J.-F.; Crowe, L.; de Vito, G.; O’Shea, D.; Caulfield, B. Comparative effect of a 1 h session of electrical muscle stimulation and walking activity on energy expenditure and substrate oxidation in obese subjects. Appl. Physiol. Nutr. Metab. 2013, 38, 57–65.
- Nieuwoudt, S.; Mulya, A.; Fealy, C.E.; Martelli, E.; Dasarathy, S.; Prasad, S.V.N.; Kirwan, J.P. In vitro contraction protects against palmitate-induced insulin resistance in C2C12 myotubes. Am. J. Physiol. Physiol. 2017, 313, C575–C583.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
693
Revisions:
2 times
(View History)
Update Date:
13 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No