Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Quancan Hou | + 3281 word(s) | 3281 | 2021-12-07 02:21:16 |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hou, Q. Epigenome and Epitranscriptome: Potential Resources for Crop Improvement. Encyclopedia. Available online: https://encyclopedia.pub/entry/16976 (accessed on 28 June 2026).
Hou Q. Epigenome and Epitranscriptome: Potential Resources for Crop Improvement. Encyclopedia. Available at: https://encyclopedia.pub/entry/16976. Accessed June 28, 2026.
Hou, Quancan. "Epigenome and Epitranscriptome: Potential Resources for Crop Improvement" Encyclopedia, https://encyclopedia.pub/entry/16976 (accessed June 28, 2026).
Hou, Q. (2021, December 10). Epigenome and Epitranscriptome: Potential Resources for Crop Improvement. In Encyclopedia. https://encyclopedia.pub/entry/16976
Hou, Quancan. "Epigenome and Epitranscriptome: Potential Resources for Crop Improvement." Encyclopedia. Web. 10 December, 2021.
Copy Citation
Crop breeding faces the challenge of increasing food demand, especially under climatic changes. Conventional breeding has relied on genetic diversity by combining alleles to obtain desired traits. In recent years, research on epigenetics and epitranscriptomics has shown that epigenetic and epitranscriptomic diversity provides additional sources for crop breeding and harnessing epigenetic and epitranscriptomic regulation through biotechnologies has great potential for crop improvement.
epigenetics
epitranscriptomics
epigenome editing
epitranscriptome engineering
crop improvement
1. Introduction
Since the birth of agriculture, human beings have never stopped domesticating plants. For thousands of years, we have selectively bred crops with desirable traits, such as high yield, nutritious, biotic- and abiotic resistance, etc. Most modern crop varieties, including rice (Oryza sativa), wheat (Triticum aestivum), and maize (Zea mays), are obtained from conventional breeding approaches, which rely on the selection and collection of favorable alleles from the offspring of crossed varieties. Although modern varieties provide nutritious crops with high yields, the global human population is predicted to reach 10 billion by 2050 and will exceed our ability to meet the nutritional needs of humans around the world [1].
Breeders and plant scientists have been applying different strategies to accelerate the breeding process. For example, by extending photoperiods and controlling temperatures, the so-called “speed breeding”, the generation times of wheat, barley (Hordeum vulgare), chickpea (Cicer arietinum), pea (Pisum sativum), and canola (Brassica napus) have been significantly shortened [2][3]. The explosion in available reference pangenomes allow breeders to use marker-assisted selection and genome selection easier, facilitating efficient phenotyping and genotyping plant materials [4]. The automated and machine-learning-assisted high-throughput phenotyping systems enable the efficient screening, selection, and evaluation of large populations [5][6].
Crop genetic engineering by adding or editing genetic information can increase yield and improve crops in adverse environments. For instance, overexpression of OsDREB genes leads to enhanced drought tolerance in rice [7]. Higher expression of OsIPA1 by overexpression or mutation at the miR156 and miR529 target sites has improved grain yield and immunity in rice [8]. Increased OsGRF4 abundance elevates grain yields of rice and wheat grown in moderate nitrogen-supply [9]. Genetic variation generated through genome editing such as CRISPR/Cas can be indistinguishable from naturally occurring variation and thus should be readily accessible for commercialization. Using the CRISPR-Cas9 system, multiple endogenous genes that function in plant architecture, plant immunity, nitrogen use, and other pathways have been manipulated to improve crops directly. Many genes have been targeted by using genome editing platforms to engineer disease resistance [10]. Double knockout of Microrchidia MORC1 and MORC6a using CRISPR/Cas9 significantly increases the resistance of barley to biotrophic (Blumeria graminis) and necrotrophic (Fusarium graminearum) plant pathogenic fungi [11]. Knockout of FAD2 genes by CRISPR/Cas9 leads to increases in oleic acid and total monounsaturated fatty acid composition with concurrent decreases in undesirable polyunsaturated linoleic and linolenic fatty acid content [12]. CRISPR/Cas9-mediated gene editing of GmJAGGED1 increased yield in a low-latitude soybean variety [13]. With the help of genome editing, a remarkable work to domesticate wild allotetraploid rice de novo into a new staple cereal has been reported recently. Six agronomically important traits were rapidly improved by editing O. alta homologs of the genes controlling these traits in diploid rice [14]. Such strategies described above will greatly accelerate the breeding process and strengthen world food security.
Thus far, plant breeding has made use of genetic variation, but epigenetic factors, such as DNA methylation, can also be heritable and can contribute to breeding. Epigenetics is the study of heritable changes in genome function that are not attributed to alterations of the DNA sequence but involve the control of DNA packaging to switch genes on or off. In plants, many biological processes are associated with epigenetic regulation, such as vernalization, paramutation, transgenic silencing, imprinting, etc. Epitranscriptomics has revealed that RNA modifications are critical posttranscriptional regulators of gene expression affecting that cell differentiation and development [15]. Knowledge on epigenetic and epitranscriptomic control for plant development and biotic and abiotic resistance is accumulating, and epigenetic and epitranscriptomic editing for crop breeding is emerging [16][17][18][19].
2. Epigenetics and Epitranscriptomics
Epigenetic mechanisms play essential roles in all kingdoms of life and these mechanisms generally include DNA and histone modifications, histone variants, and some non-coding RNAs (ncRNAs) [20] (Figure 1). It is known that each of the four DNA bases could be chemically modified and at least 17 DNA modifications have been discovered, among which 5-methylcytosine (5mC) is the best characterized [21]. In plants, de novo DNA methylation is established by the RNA-directed DNA methylation (RdDM) pathway and DNA methylation on the sequence contexts CG, CHG (where H = A, C or T), and CHH is maintained by different DNA methyltransferases [22]. 5mC can be actively removed by 5-methylcytosine DNA demethylases, a kind of DNA glycosylase/lyase family enzymes [22]. 5mC is dynamically regulated and tightly associated with other chromatin elements, exerting widespread effects on gene expression during plant development and in response to environmental factors. The effects highly depend on the location of the methylation relative to the gene. 5mC present over the transcription start site often leads to gene silencing while the gene body 5mC has minimal effects on gene expression [23][24]. Recently, N6-methyladenine (6mA) modification has also been identified as a new epigenetic mark in plants [25][26][27]. Unlike the silencing function of 5mC in gene promoters, the distribution of 6mA on genomes is divergent among species and its effect needs to be investigated. Though information on 6mA is less known, the available evidence suggests that it functions in plant development, tissue differentiation, and gene expression regulation [26][27].
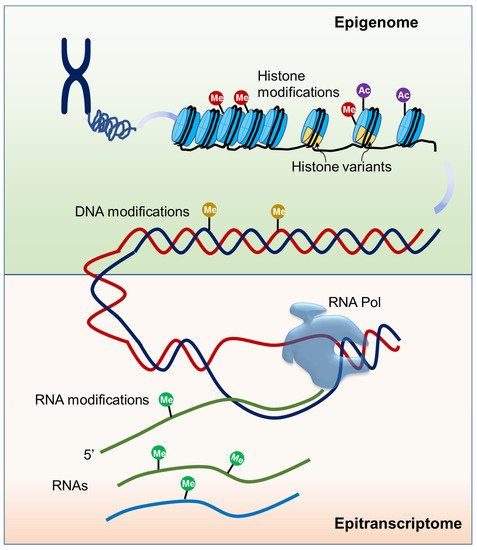
Figure 1. Schematic of epigenome and epitranscriptome. Epigenome is mainly composed of modifications of DNA and histone proteins. Epitranscriptome is composed of all biochemical RNA modifications.
Modifications at histone residues mainly include methylation, acetylation, phosphorylation, and ubiquitination. These covalent modifications on histones called the “histone code” can alter chromatin structure or recruit interaction effectors, influencing transcriptional activity. The different types of histone modifications play different roles in specifying chromatin function. For example, histone H3 with tri-methylation on lysine 4 (H3K4me3) and 36 (H3K36me3) is often distributed on actively expressed genes and associated with euchromatin, whereas H3K27me1 and H3K9me2 are usually present within heterochromatic regions [28]. In addition to histone methylation, other histone marks such as acetylation, phosphorylation, and ubiquitination are also associated with gene expression regulation. Acetylation can occur at many lysine residues of H2A, H2B, H3, and H4 [29]. Histone acetylation relaxes the chromatin structure and leads to transcriptional activation, while histone deacetylation condenses the chromatin structure, often resulting in transcriptional repression [30]. Histone marks are established, recognized, and removed by specific proteins or protein complexes that are referred to as the writers, readers, and erasers, respectively [28][31]. For example, H3K4me3 deposition is catalyzed by the methyltransferases Arabidopsis Trithoras-like Protein1 (ATX1) and ATX2 in Arabidopsis [32]. H3K27me3 is catalyzed by the polycomb repressive complex 2 (PRC2) via the histone methyltransferases Curly Leaf (CLF), Swinger (SWN), and Medea (MEA), and can be recognized by the PRC1 complex through the reader proteins Like Heterochromatin Protein 1 (LHP1), Early Bolting in Short Day (EBS), and Short Life (SHL) [33][34]. The removal of histone lysine methylation is catalyzed by jumonji C (JmjC) domain-containing proteins and lysine-specific demethylase1 (LSD1)-like proteins [28].
In addition to DNA and histone modifications, three main types of RNA are also subjected to biochemical modifications (Figure 1). Although it was revealed long ago that chemical modifications are critical for ncRNAs to facilitate their full function, modifications on mRNA are recently disclosed to be important for RNA metabolism [35]. So far, about 160 chemical modifications have been discovered in RNA, and N6-methyladenosine (m6A) is one of the most abundant modifications on mRNA. m6A amount is estimated to account for 0.1–0.4% of the total adenosine in cellular mRNA, approximately 2–3 sites per transcript [36]. m6A mRNA modification is catalyzed by a conserved multicomponent methyltransferase complex in eukaryotes. In Arabidopsis, mRNA adenosine methyltransferase MTA, MTB, Fkbp12 Interacting Protein 37KD (FIP37), Kiaa1229/Virlizer (VIR), and Hakai have been reported to have adenosine methyltransferase activity, and knockout or knockdown any of these factors result in decreased m6A levels [37][38][39][40]. ALKBH family proteins were identified as m6A demethylases to remove methyl groups [41][42]. YTH domain-containing proteins were identified as reader proteins that bind to m6A-modified mRNA in vivo and affect mRNA stability in Arabidopsis [43]. Another mRNA modification, 5-methylcytosine (m5C), has been detected in different eukaryotes, including Arabidopsis [44]. The distribution of m5C on mRNA is still unclear. RNA bisulfite sequencing (RNA-BisSeq) analysis revealed that m5C is abundant in 3’UTRs while m5C-RIP-seq analysis showed that m5C is enriched in coding sequence [44][45]. RNA (C5-cytosine) methyltransferase (RCMT) family proteins have been identified as m5C mRNA methyltransferases in Arabidopsis. So far, there has been no m5C-binding protein identified in plants [46]. Recently, the N4-acetylcytidine modification (ac4C) has been identified as a new reversible RNA modification present in tRNA, rRNA, and mRNA, and plays a vital role in mRNA stability and translation fidelity [47]. In humans, ac4C is catalyzed by the N-acetyltransferase 10, and SIRT7 has been identified as a deacetylase [48]. However, nothing is known about ac4C mRNA modification in plants. Several other RNA modifications such as N6,2′-O-dimethyladenosine (m6Am), 8-oxo-7,8-dihydroguanosine (8-oxoG), and pseudouridine (Ψ) have also been shown to influence the mRNA stability and consequently affect translation efficiency [49].
3. Epigenomic and Epitranscriptomic Changes during Development
Much evidence has indicated that epigenetic and epitranscriptomic modification profiles vary in plant-specific organs and cell types. DNA methylation in the CHH context displays significant differences among leaves, flowers, and ovules, in line with small RNA abundance at corresponding sites [50]. The DNA methylation variations could be partially attributed to the tissue-specific expression of Classy (CLSY) genes, which encode chromatin remodelers that are involved in the RNA-directed DNA methylation (RdDM) pathway by facilitating RNA polymerase IV (Pol IV) recruitment and small RNA generation [50]. A comparison of DNA 5mC methylomes of the shoot apical meristem revealed that CHG methylation and CHH methylation were increased after the transition from vegetative to reproductive growth in Arabidopsis and rice, respectively [51][52]. Although most root cell types have similar 5mC landscapes, columella displayed genome-wide hypermethylation in the CHH context [53]. The increased mCHH in columella is mainly distributed in transposable elements, and this might be a mechanism to keep the neighboring stem cells silenced during the root development [54]. Several studies identified 5mC changes in the male reproductive cells [55][56][57]. Some regions gain methylation in the sex cells in the CHH context via RdDM, and genes within these regions are upregulated in an RdDM mutant in meiocytes but not in leaves. This suggests RdDM is required for the silencing of these genes, specifically in the male sex lineage [57]. Transposable elements (TEs) have reduced 5mC DNA methylation in the vegetative nucleus (VN) but not in sperm cells (SC), resulting in the generation of 21 nucleotide siRNAs from Athila retrotransposons in VN. The VN-generated siRNAs could further target TEs in gametes and ensure gamete TEs are silenced, which is essential for the silencing of TE in the next generation [55]. Similarly, the meiocytes’ nurse cells generate TE-derived small RNAs that can distribute into meiocytes and lead to TE silencing by RdDM [56]. DNA methylation alteration in gametes could be significant for the inheritance of DNA methylation and may provide potential targets for generating DNA methylation variation in crop species. In addition, DNA methylome alterations have been documented in soybean development and during tomato fruit ripening [58][59]. The level of 6mA also shows a dynamic pattern in plant development. In Arabidopsis, 6mA accumulation during vegetative development is significantly correlated with the upregulation of gene expression [26]. In rice, the mutation of Deficient in DNA Methylation 1 (DDM1) that significantly decreased the level of 6mA resulted in downregulation of gene expression [27]. These results suggest 6mA is associated with actively expressed genes, which is in contrast to 5mC. However, 6mA is also enriched on transposable elements and over the pericentromeric heterochromatin regions [26][27].
Some histone marks also show dynamic changes in plant development. For example, the profile of H3K27me3 varies among different tissues of maize [60], and genes of Arabidopsis are differentially marked by H3K27me3 during cell type transitions [61]. Knockout of components of the polycomb group (PcG) chromatin remodeling complex responsible for catalyzation of H3K27me3 results in abnormal development [62], suggesting H3K27me3 plays an essential role in defining plant cell fate. H3K27me3, H3K4me3, and gene expression profiling in Arabidopsis in different root cells and guard cells demonstrated that H3K27me3 dynamics regulate cell identity [61][63]. A comparison between young and mature leaves revealed a relationship between gene expression changes and H3.3 content on the affected genes [64].
Measurement of the level of mRNA modifications by using different approaches revealed that mRNA modifications display dynamic patterns in plant development. In Arabidopsis, transcriptome-wide m6A-seq revealed that 33.5% of transcripts showed differential m6A methylation between leaves, flowers, and roots [65]. Thin-layer chromatography analysis showed m6A levels differ among different tissues, with a high ratio (1.5%) in young seedlings and relatively lower ratios in leaf (0.9%) and root (0.6%) [37]. Analysis by liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) revealed m5C levels ranged from 0.01% in rosette leaves to 0.036% in siliques, with m5C abundance slightly increasing from 3-day-old (0.027%) to 15-day-old (0.033%) seedlings [44]. RNA bisulfite sequencing of siliques, shoots, and roots tissues of Arabidopsis showed that most m5C sites were tissue specific, and only 15 sites were commonly methylated between all three tissue types [45]. In rice, 1792 and 6508 tissue-specific m6A-modified genes were identified in callus and leaves, respectively [66]. Dynamic changes of mRNA m6A modification have also been observed during tomato fruit ripening [67]. Consistently, the transcript levels of writer, eraser, and reader coding genes vary in different tissues and during plant development [37][38][44]. Disruption of the components of writers, erasers, or readers would alter mRNA decay rates and often cause severe developmental problems. For example, the knockout of the genes encoding core m6A writer components results in embryonic lethality [37][68]. Loss of function of the m6A reader ECT2 affects mRNA stability degradation of the trichome development-related transcripts and leads to more extensively branched trichomes [43]. Mutations in TRM4B, encoding an m5C methyltransferase, display primary and lateral root development defects and decreased m5C levels on root development-related genes [44].
4. Epigenome and Epitranscriptome Engineering for Crop Improvement
Recently, epigenome editing tools that specifically target a genome locus to change epigenetic modifications (cytosine de/methylation or histone tail de/methylation, de/acetylation, etc.) have been developed, enabling precise generation of artificial epialleles. These approaches were designed by fusing epigenetic modifiers or an interacting platform that can recruit the epimodifiers to nuclease-deficient genome editing tools, which guides the fused functional module to a predefined site and directly cause localized epigenome changes (Figure 2A,B). The zinc-finger (ZF) protein, transcription activator-like effector protein, and nuclease-dead CRISPR-associated protein 9 (dCas9) were commonly used for specific DNA sequence targeting. A chemically inducible dCas9 system has been successfully used in human cells [69] (Figure 2C), and a light-inducible dCas9 system was proposed to be adapted for epigenome editing [70] (Figure 2D). Successful applications of these epigenome editing tools have been shown at the FWA locus in Arabidopsis. The Flowering Wageningen (FWA) is a flowering repressor, and its promoter has tandem repeats that can be methylated or demethylated, resulting in gene silencing or activation, respectively [71]. The demethylated epiallele fwa displayed a delayed flowering phenotype. The ZF protein fused with RdDM components such as SUVH2, SHH1, NRPD1, RDR2, DMS3, or RDM and directed to the FWA promoter induces DNA methylation at the target sites [72][73]. Interestingly, co-targeting of ZF–DMS3 and ZF–NRPD1 enhanced the targeted methylation, suggesting multiple silencing factors have a synergistic effect when they are simultaneously recruited to a defined site [73]. Fusing a ZF or a dCas9 with the catalytic domain of the human DNA demethylase TET1 also led to efficient demethylation of the targeted FWA promoter [74]. Induced methylation or demethylation at the FWA promoter, which results in the creation of early or late phenotypes, are heritable traits, even when the epigenome editing module was segregated away, suggesting the stable creation of the epialleles [75]. Besides the FWA locus, when the fusion protein ZF-TET1 targeted the methylated regions of the CACTA1 transposon, this also resulted in targeted demethylation and changes in expression [74].
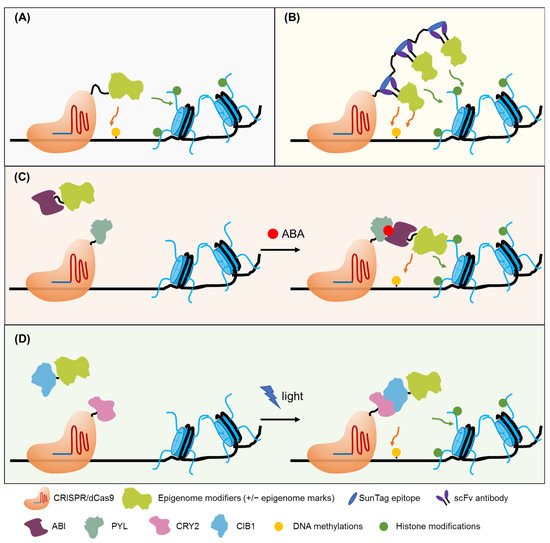
Figure 2. Epigenome editing tools. (A) Direct epigenome editing. Fusions of epigenome modifiers to deactivated Cas9 (dCas9) can be directed to specific loci and cause epigenetic changes of interest. (B) Enhanced epigenome editing. dCas9 is fused to SunTag epitopes and the single-chain variable fragment (scFv) is fused to epigenome modifiers. Multiple copies of scFv-epigenome modifiers can be directed to specific loci and cause epigenetic changes of interest. (C) Chemically inducible epigenome editing. ABA mediates the interaction of ABI and PYL to direct epigenome to dCas9-gRNA-targeting sites for epigenome editing. (D) Light-inducible epigenome editing. Light induces the interaction of CRY2 and CIB1 to direct epigenome modifiers to dCas9-gRNA-targeting sites for epigenome editing.
Some RNA modifications have indispensable roles in plant development and tolerance to various environmental stresses, which are closely associated with agricultural traits [76]. For instance, m6A mRNA modification regulates strawberry fruit ripening [77]. OsNSUN2-mediated m5C mRNA modification has been shown to enhance rice adaptation to high-temperature stress [78]. m6A mRNA modification plays a vital role in salt-stress tolerance in Arabidopsis [79]. Thus, epitranscriptome manipulation has great potential for improving crop traits. Recent studies revealed that harnessing m6A regulation could remarkably improve economically important traits in crops [16][77][80]. Transgenic expression of a human RNA demethylase FTO (fat mass and obesity associated) in rice and potato stimulates root meristem cell proliferation, tiller bud formation, promotes photosynthetic efficiency, and results in ~50% increases in yield and biomass. Mechanistically, FTO causes substantial m6A demethylation of both mRNA and repeat RNA in the transgenic plants. m6A demethylation of plant repeat RNA further induces chromatin openness and subsequently causes a global transcriptional upregulation of tissue-specific genes encoding proteins that play functional roles in root cell proliferation, tiller formation, and photosynthetic efficiency [16]. Overexpression of PtrMTA encoding a component of the m6A methyltransferase complex that participated in the formation of m6A methylation exhibits enhanced poplar tolerance to drought stress. Poplar plants that overexpression of PtrMTA displayed an increased density of trichomes and a more developed root system than that of the wild type [80]. In strawberry, the overexpression of FveMTA or FveMTB, encoding m6A methyltransferases, accelerates fruit ripening, while the suppression of either delays fruit ripening, providing a good example of fruit maturity control through epitranscriptome manipulation [77]. Strategies for epitranscriptome engineering have been proposed from different angles (Figure 3): (1) Manipulating the activities of RNA modification-related proteins, including writers, readers, and erasers, by generating gain-of-function or loss-of-function mutants [81]; (2) specific RNA editing using fusions of catalytically inactivated dCas13 and RNA modification enzymes to create or remove RNA modifications on target sites [82]; and (3) eliminating specific RNA modification by manipulation at the DNA level, which requires precise base editors to generate synonymous mutation [83]. So far, only the first strategy has been applied in plants. The prerequisite for applying the other strategies needs a comprehensive understanding of the epitranscriptome at a single-base resolution and the associations with phenotypic outputs.
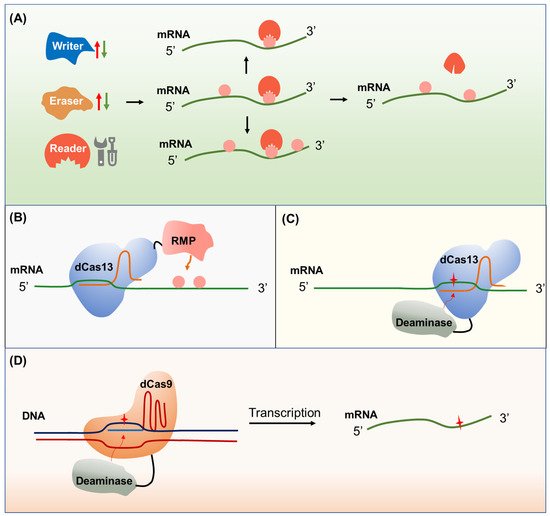
Figure 3. Epitranscriptome engineering tools. (A) Modulating the activity of RNA modification proteins by promoting or inhibiting the RNA modification writer or eraser proteins or manipulating the RNA modification reader proteins to trigger global RNA modification changes. (B) Direct epitranscriptome editing. Fusions of RNA modification proteins (RMP) to deactivated Cas13 (dCas13) can be directed to specific transcripts and cause epigenetic changes of interest. (C) Epitranscriptome editing through RNA base editing. Fusions of deaminase to deactivated dCas13 can be directed to the specific transcript for RNA base editing. The resulting synonymous mutations might cause RNA modification changes. (D) Epitranscriptome editing through DNA base editing. Fusions of deaminase to dCas9 can be directed to a specific locus for DNA base editing. The resulting synonymous mutations might further cause RNA modification changes.
References
- Hickey, L.T.; Hafeez, A.N.; Robinson, H.; Jackson, S.A.; Leal-Bertioli, S.C.M.; Tester, M.; Gao, C.; Godwin, I.D.; Hayes, B.J.; Wulff, B.B.H. Breeding crops to feed 10 billion. Nat. Biotechnol. 2019, 37, 744–754.
- Watson, A.; Ghosh, S.; Williams, M.J.; Cuddy, W.S.; Simmonds, J.; Rey, M.-D.; Hatta, M.A.M.; Hinchliffe, A.; Steed, A.; Reynolds, D.; et al. Speed breeding is a powerful tool to accelerate crop research and breeding. Nat. Plants 2018, 4, 23–29.
- Song, Y.; Duan, X.; Wang, P.; Li, X.; Yuan, X.; Wang, Z.; Wan, L.; Yang, G.; Hong, D. Comprehensive speed breeding: A high-throughput and rapid generation system for long-day crops. Plant Biotechnol. J. 2021.
- Xu, Y.; Crouch, J.H. Marker-Assisted Selection in Plant Breeding: From Publications to Practice. Crop. Sci. 2008, 48, 391–407.
- Araus, J.L.; Kefauver, S.C.; Zaman-Allah, M.; Olsen, M.S.; Cairns, J. Translating High-Throughput Phenotyping into Genetic Gain. Trends Plant Sci. 2018, 23, 451–466.
- Newman, S.J.; Furbank, R.T. Explainable machine learning models of major crop traits from satellite-monitored continent-wide field trial data. Nat. Plants 2021, 7, 1354–1363.
- Chen, J.-Q.; Meng, X.-P.; Zhang, Y.; Xia, M.; Wang, X.-P. Over-expression of OsDREB genes lead to enhanced drought tolerance in rice. Biotechnol. Lett. 2008, 30, 2191–2198.
- Wang, J.; Zhou, L.; Shi, H.; Chern, M.; Yu, H.; Yi, H.; He, M.; Yin, J.; Zhu, X.; Li, Y.; et al. A single transcription factor promotes both yield and immunity in rice. Science 2018, 361, 1026–1028.
- Li, S.; Tian, Y.; Wu, K.; Ye, Y.; Yu, J.; Zhang, J.; Liu, Q.; Hu, M.; Li, H.; Tong, Y.; et al. Modulating plant growth–metabolism coordination for sustainable agriculture. Nature 2018, 560, 595–600.
- Zaidi, S.S.E.A.; Mukhtar, S.; Mansoor, S. Genome Editing: Targeting Susceptibility Genes for Plant Disease Resistance. Trends Biotechnol. 2018, 36, 898–906.
- Galli, M.; Martiny, E.; Imani, J.; Kumar, N.; Koch, A.; Steinbrenner, J.; Kogel, K.-H. CRISPR/SpCas9-mediated double knockout of barley Microrchidia MORC1 and MORC6a reveals their strong involvement in plant immunity, transcriptional gene silencing and plant growth. Plant Biotechnol. J. 2021.
- Jiang, W.Z.; Henry, I.M.; Lynagh, P.G.; Comai, L.; Cahoon, E.B.; Weeks, D.P. Significant enhancement of fatty acid composition in seeds of the allohexaploid, Camelina sativa, using CRISPR /Cas9 gene editing. Plant Biotechnol. J. 2017, 15, 648–657.
- Cai, Z.; Xian, P.; Cheng, Y.; Ma, Q.; Lian, T.; Nian, H.; Ge, L. CRISPR/Cas9-mediated gene editing of GmJAGGED1 increased yield in the low-latitude soybean variety Huachun 6. Plant Biotechnol. J. 2021, 19, 1898–1900.
- Yu, H.; Lin, T.; Meng, X.; Du, H.; Zhang, J.; Liu, G.; Chen, M.; Jing, Y.; Kou, L.; Li, X.; et al. A route to de novo domestication of wild allotetraploid rice. Cell 2021, 184, 1156–1170.
- Frye, M.; Harada, B.T.; Behm, M.; He, C. RNA modifications modulate gene expression during development. Science 2018, 361, 1346–1349.
- Yu, Q.; Liu, S.; Yu, L.; Xiao, Y.; Zhang, S.; Wang, X.; Xu, Y.; Yu, H.; Li, Y.; Yang, J.; et al. RNA demethylation increases the yield and biomass of rice and potato plants in field trials. Nat. Biotechnol. 2021, 1–8.
- Wang, Z.; Chen, D.; Sun, F.; Guo, W.; Wang, W.; Li, X.; Lan, Y.; Du, L.; Li, S.; Fan, Y.; et al. ARGONAUTE 2 increases rice susceptibility to rice black-streaked dwarf virus infection by epigenetically regulating HEXOKINASE 1 expression. Mol. Plant Pathol. 2021, 22, 1029–1040.
- Gao, Q.; Zhang, N.; Wang, W.-Q.; Shen, S.-Y.; Bai, C.; Song, X.-J. The ubiquitin-interacting motif-type ubiquitin receptor HDR3 interacts with and stabilizes the histone acetyltransferase GW6a to control the grain size in rice. Plant Cell 2021, 33, 3331–3347.
- Habig, M.; Lorrain, C.; Feurtey, A.; Komluski, J.; Stukenbrock, E.H. Epigenetic modifications affect the rate of spontaneous mutations in a pathogenic fungus. Nat. Commun. 2021, 12, 1–13.
- Wang, J.; Meng, X.; Yuan, C.; Harrison, A.P.; Chen, M. The roles of cross-talk epigenetic patterns inArabidopsis thaliana. Brief. Funct. Genom. 2016, 15, 278–287.
- Raiber, E.-A.; Hardisty, R.; van Delft, P.; Balasubramanian, S. Mapping and elucidating the function of modified bases in DNA. Nat. Rev. Chem. 2017, 1, 69.
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506.
- Niederhuth, C.E.; Bewick, A.J.; Ji, L.; Alabady, M.S.; Kim, K.D.; Li, Q.; Rohr, N.A.; Rambani, A.; Burke, J.M.; Udall, J.A.; et al. Widespread natural variation of DNA methylation within angiosperms. Genome Biol. 2016, 17, 194.
- Bewick, A.J.; Ji, L.; Niederhuth, C.E.; Willing, E.-M.; Hofmeister, B.T.; Shi, X.; Wang, L.; Lu, Z.; Rohr, N.A.; Hartwig, B.; et al. On the origin and evolutionary consequences of gene body DNA methylation. Proc. Natl. Acad. Sci. USA 2016, 113, 9111–9116.
- Zhou, C.; Wang, C.; Liu, H.; Zhou, Q.; Liu, Q.; Guo, Y.; Peng, T.; Song, J.; Zhang, J.; Chen, L.; et al. Identification and analysis of adenine N6-methylation sites in the rice genome. Nat. Plants 2018, 4, 554–563.
- Liang, Z.; Shen, L.; Cui, X.; Bao, S.; Geng, Y.; Yu, G.; Liang, F.; Xie, S.; Lu, T.; Gu, X.; et al. DNA N6-Adenine Methylation in Arabidopsis thaliana. Dev. Cell 2018, 45, 406–416.
- Zhang, Q.; Liang, Z.; Cui, X.; Ji, C.; Li, Y.; Zhang, P.; Liu, J.; Riaz, A.; Yao, P.; Liu, M.; et al. N6-Methyladenine DNA Methylation in Japonica and Indica Rice Genomes and Its Association with Gene Expression, Plant Development, and Stress Responses. Mol. Plant 2018, 11, 1492–1508.
- Liu, C.; Lu, F.; Cui, X.; Cao, X. Histone Methylation in Higher Plants. Annu. Rev. Plant Biol. 2010, 61, 395–420.
- Zhang, K.; Sridhar, V.V.; Zhu, J.; Kapoor, A.; Zhu, J.-K. Distinctive Core Histone Post-Translational Modification Patterns in Arabidopsis thaliana. PLoS ONE 2007, 2, e1210.
- Shahbazian, M.D.; Grunstein, M. Functions of Site-Specific Histone Acetylation and Deacetylation. Annu. Rev. Biochem. 2007, 76, 75–100.
- Marmorstein, R.; Zhou, M.-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762.
- Saleh, A.; Alvarez-Venegas, R.; Yilmaz, M.; Le, O.; Hou, G.; Sadder, M.; Al-Abdallat, A.; Xia, Y.; Lu, G.; Ladunga, I.; et al. The Highly Similar Arabidopsis Homologs of Trithorax ATX1 and ATX2 Encode Proteins with Divergent Biochemical Functions. Plant Cell 2008, 20, 568–579.
- Mozgova, I.; Hennig, L. The Polycomb Group Protein Regulatory Network. Annu. Rev. Plant Biol. 2015, 66, 269–296.
- Yang, Z.; Qian, S.; Scheid, R.N.; Lu, L.; Chen, X.; Liu, R.; Du, X.; Lv, X.; Boersma, M.D.; Scalf, M.; et al. EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat. Genet. 2018, 50, 1247–1253.
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650.
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646.
- Zhong, S.; Li, H.; Bodi, Z.; Button, J.; Vespa, L.; Herzog, M.; Fray, R.G. MTA Is an Arabidopsis Messenger RNA Adenosine Methylase and Interacts with a Homolog of a Sex-Specific Splicing Factor. Plant Cell 2008, 20, 1278–1288.
- Shen, L.; Liang, Z.; Gu, X.; Chen, Y.; Teo, Z.W.N.; Hou, X.; Cai, W.M.; Dedon, P.C.; Liu, L.; Yu, H. N6-Methyladenosine RNA Modification Regulates Shoot Stem Cell Fate in Arabidopsis. Dev. Cell 2016, 38, 186–200.
- Růžička, K.; Zhang, M.; Campilho, A.; Bodi, Z.; Kashif, M.; Saleh, M.; Eeckhout, D.; El-Showk, S.; Li, H.; Zhong, S.; et al. Identification of factors required for m 6 A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. N. Phytol. 2017, 215, 157–172.
- Zhang, F.; Zhang, Y.-C.; Liao, J.-Y.; Yu, Y.; Zhou, Y.-F.; Feng, Y.-Z.; Yang, Y.-W.; Lei, M.-Q.; Bai, M.; Wu, H.; et al. The subunit of RNA N6-methyladenosine methyltransferase OsFIP regulates early degeneration of microspores in rice. PLoS Genet. 2019, 15, e1008120.
- Duan, H.-C.; Wei, L.-H.; Zhang, C.; Wang, Y.; Chen, L.; Lu, Z.; Chen, P.R.; He, C.; Jia, G. ALKBH10B Is an RNA N6-Methyladenosine Demethylase Affecting Arabidopsis Floral Transition. Plant Cell 2017, 29, 2995–3011.
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760.
- Wei, L.-H.; Song, P.; Wang, Y.; Lu, Z.; Tang, Q.; Yu, Q.; Xiao, Y.; Zhang, X.; Duan, H.-C.; Jia, G. The m6A Reader ECT2 Controls Trichome Morphology by Affecting mRNA Stability in Arabidopsis. Plant Cell 2018, 30, 968–985.
- Cui, X.; Liang, Z.; Shen, L.; Zhang, Q.; Bao, S.; Geng, Y.; Zhang, B.; Leo, V.; Vardy, L.; Lu, T.; et al. 5-Methylcytosine RNA Methylation in Arabidopsis Thaliana. Mol. Plant 2017, 10, 1387–1399.
- David, R.; Burgess, A.; Parker, B.; Li, J.; Pulsford, K.; Sibbritt, T.; Preiss, T.; Searle, I.R. Transcriptome-Wide Mapping of RNA 5-Methylcytosine in Arabidopsis mRNAs and Noncoding RNAs. Plant Cell 2017, 29, 445–460.
- Liang, Z.; Riaz, A.; Chachar, S.; Ding, Y.; Du, H.; Gu, X. Epigenetic Modifications of mRNA and DNA in Plants. Mol. Plant 2020, 13, 14–30.
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886.
- Kudrin, P.; Meierhofer, D.; Vågbø, C.B.; Ørom, U.A.V. Nuclear RNA-acetylation can be erased by the deacetylase SIRT7. bioRxiv 2021.
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408.
- Zhou, M.; Coruh, C.; Xu, G.; Bourbousse, C.; Lambolez, A.; Law, J.A. The CLASSY family controls tissue-specific DNA methylation patterns in Arabidopsis. bioRxiv 2021.
- Gutzat, R.; Rembart, K.; Nussbaumer, T.; Hofmann, F.; Pisupati, R.; Bradamante, G.; Daubel, N.; Gaidora, A.; Lettner, N.; Donà, M.; et al. Arabidopsis shoot stem cells display dynamic transcription and DNA methylation patterns. EMBO J. 2020, 39, 103667.
- Higo, A.; Saihara, N.; Miura, F.; Higashi, Y.; Yamada, M.; Tamaki, S.; Ito, T.; Tarutani, Y.; Sakamoto, T.; Fujiwara, M.; et al. DNA methylation is reconfigured at the onset of reproduction in rice shoot apical meristem. Nat. Commun. 2020, 11, 1–12.
- Kawakatsu, T.; Stuart, T.; Valdes, M.; Breakfield, N.; Schmitz, R.; Nery, J.R.; Urich, M.A.; Han, X.; Lister, R.; Benfey, P.N.; et al. Unique cell-type-specific patterns of DNA methylation in the root meristem. Nat. Plants 2016, 2, 1–8.
- Lloyd, J.P.B.; Lister, R. Epigenome plasticity in plants. Nat. Rev. Genet. 2021, 1–14.
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdžić, M.; Becker, J.D.; Feijó, J.A.; Martienssen, R.A. Epigenetic Reprogramming and Small RNA Silencing of Transposable Elements in Pollen. Cell 2009, 136, 461–472.
- Long, J.; Walker, J.; She, W.; Aldridge, B.; Gao, H.; Deans, S.; Vickers, M.; Feng, X. Nurse cell –derived small RNAs define paternal epigenetic inheritance in Arabidopsis. Science 2021, 373, 556.
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137.
- Lang, Z.; Wang, Y.; Tang, K.; Tang, D.; Datsenka, T.; Cheng, J.; Zhang, Y.; Handa, A.K.; Zhu, J.-K. Critical roles of DNA demethylation in the activation of ripening-induced genes and inhibition of ripening-repressed genes in tomato fruit. Proc. Natl. Acad. Sci. USA 2017, 114, E4511–E4519.
- Song, Q.-X.; Lu, X.; Li, Q.-T.; Chen, H.; Hu, X.-Y.; Ma, B.; Zhang, W.-K.; Chen, S.-Y.; Zhang, J.-S. Genome-Wide Analysis of DNA Methylation in Soybean. Mol. Plant 2013, 6, 1961–1974.
- Makarevitch, I.; Eichten, S.; Briskine, R.; Waters, A.J.; Danilevskaya, O.N.; Meeley, R.B.; Myers, C.L.; Vaughn, M.; Springer, N.M. Genomic Distribution of Maize Facultative Heterochromatin Marked by Trimethylation of H3K27. Plant Cell 2013, 25, 780–793.
- Lee, L.; Wengier, D.L.; Bergmann, D.C. Cell-type–specific transcriptome and histone modification dynamics during cellular reprogramming in the Arabidopsis stomatal lineage. Proc. Natl. Acad. Sci. USA 2019, 116, 21914–21924.
- Ikeuchi, M.; Iwase, A.; Rymen, B.; Harashima, H.; Shibata, M.; Ohnuma, M.; Breuer, C.; Morao, A.K.; De Lucas, M.; De Veylder, L.; et al. PRC2 represses dedifferentiation of mature somatic cells in Arabidopsis. Nat. Plants 2015, 1, 15089.
- Deal, R.B.; Henikoff, S. A simple method for gene expression and chromatin profiling of individual cell types within a tissue. Dev. Cell 2010, 18, 1030–1040.
- Wollmann, H.; Holec, S.; Alden, K.; Clarke, N.D.; Jacques, P.-E.; Berger, F. Dynamic Deposition of Histone Variant H3.3 Accompanies Developmental Remodeling of the Arabidopsis Transcriptome. PLoS Genet. 2012, 8, e1002658.
- Wan, Y.; Tang, K.; Zhang, D.; Xie, S.; Zhu, X.; Wang, Z.; Lang, Z. Transcriptome-wide high-throughput deep m6A-seq reveals unique differential m6A methylation patterns between three organs in Arabidopsis thaliana. Genome Biol. 2015, 16, 1–26.
- Li, Y.; Wang, X.; Li, C.; Hu, S.; Yu, J.; Song, S. Transcriptome-wide N6-methyladenosine profiling of rice callus and leaf reveals the presence of tissue-specific competitors involved in selective mRNA modification. RNA Biol. 2014, 11, 1180–1188.
- Zhou, L.; Tian, S.; Qin, G. RNA methylomes reveal the m6A-mediated regulation of DNA demethylase gene SlDML2 in tomato fruit ripening. Genome Biol. 2019, 20, 1–23.
- Vespa, L.; Vachon, G.; Berger, F.; Perazza, D.; Faure, J.-D.; Herzog, M. The Immunophilin-Interacting Protein AtFIP37 from Arabidopsis Is Essential for Plant Development and Is Involved in Trichome Endoreduplication. Plant Physiol. 2004, 134, 1283–1292.
- Chen, T.; Gao, D.; Zhang, R.; Zeng, G.; Yan, H.; Lim, E.; Liang, F.-S. Chemically Controlled Epigenome Editing through an Inducible dCas9 System. J. Am. Chem. Soc. 2017, 139, 11337–11340.
- Zhao, W.; Wang, Y.; Liang, F.-S. Chemical and Light Inducible Epigenome Editing. Int. J. Mol. Sci. 2020, 21, 998.
- Soppe, W.J.J.; Jacobsen, S.E.; Alonso-Blanco, C.; Jackson, J.P.; Kakutani, T.; Koornneef, M.; Peeters, A.J.M. The Late Flowering Phenotype of fwa Mutants Is Caused by Gain-of-Function Epigenetic Alleles of a Homeodomain Gene. Mol. Cell 2000, 6, 791–802.
- Johnson, L.M.; Du, J.; Hale, C.J.; Bischof, S.; Feng, S.; Chodavarapu, R.K.; Zhong, X.; Marson, G.; Pellegrini, M.; Segal, D.J.; et al. SRA- and SET-domain-containing proteins link RNA polymerase V occupancy to DNA methylation. Nature 2014, 507, 124–128.
- Gallego-Bartolome, J.; Liu, W.; Kuo, P.H.; Feng, S.; Ghoshal, B.; Gardiner, J.; Zhao, J.M.-C.; Park, S.Y.; Chory, J.; Jacobsen, S.E. Co-targeting RNA Polymerases IV and V Promotes Efficient De Novo DNA Methylation in Arabidopsis. Cell 2019, 176, 1068–1082.
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA demethylation of the Arabidopsis genome using the human TET1 catalytic domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125.
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-specific manipulation of Arabidopsis loci using CRISPR-Cas9 SunTag systems. Nat. Commun. 2019, 10, 729.
- Shao, Y.; Wong, C.E.; Shen, L.; Yu, H. N6-methyladenosine modification underlies messenger RNA metabolism and plant development. Curr. Opin. Plant Biol. 2021, 63, 102047.
- Zhou, L.; Tang, R.; Li, X.; Tian, S.; Li, B.; Qin, G. N6-methyladenosine RNA modification regulates strawberry fruit ripening in an ABA-dependent manner. Genome Biol. 2021, 22, 168.
- Tang, Y.; Gao, C.-C.; Gao, Y.; Yang, Y.; Shi, B.; Yu, J.-L.; Lyu, C.; Sun, B.-F.; Wang, H.-L.; Xu, Y.; et al. OsNSUN2-Mediated 5-Methylcytosine mRNA Modification Enhances Rice Adaptation to High Temperature. Dev. Cell 2020, 53, 272–286.
- Hu, J.; Cai, J.; Park, S.J.; Lee, K.; Li, Y.; Chen, Y.; Yun, J.; Xu, T.; Kang, H. N 6 -Methyladenosine mRNA methylation is important for salt stress tolerance in Arabidopsis. Plant J. 2021, 106, 1759–1775.
- Lu, L.; Zhang, Y.; He, Q.; Qi, Z.; Zhang, G.; Xu, W.; Yi, T.; Wu, G.; Li, R. MTA, an RNA m6A Methyltransferase, Enhances Drought Tolerance by Regulating the Development of Trichomes and Roots in Poplar. Int. J. Mol. Sci. 2020, 21, 2462.
- Shen, L.; Yu, H. Epitranscriptome engineering in crop improvement. Mol. Plant 2021, 14, 1418–1420.
- Wilson, C.; Chen, P.J.; Miao, Z.; Liu, D.R. Programmable m6A modification of cellular RNAs with a Cas13-directed methyltransferase. Nat. Biotechnol. 2020, 38, 1431–1440.
- Kim, J.-S. Precision genome engineering through adenine and cytosine base editing. Nat. Plants 2018, 4, 148–151.
More
Information
Subjects:
Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
668
Revision:
1 time
(View History)
Update Date:
10 Dec 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No