 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sylvie GIURIATO | + 1511 word(s) | 1511 | 2021-09-24 09:00:18 | | | |
| 2 | Amina Yu | -179 word(s) | 1332 | 2021-12-10 09:41:02 | | |
Video Upload Options
Anaplastic large cell lymphoma (ALCL) is a rare type of T-cell lymphoma, accounting for 10 to 20% of childhood lymphomas. ALK-positive ALCL (ALK+ ALCL) in children carry a characteristic t(2;5) (p23;q35) chromosomal translocation, leading to the constitutive activation of the oncogenic fusion protein nucleophosmin (NPM)-ALK, which drives lymphomagenesis .
1.Introduction :
Since the current treatment of this lymphoma (mainly based on aggressive chemotherapy) is not optimal, as 30% of the patients relapse five years post-treatment[1] , considerable efforts have been made to develop therapies directly targeting the NPM-ALK oncoprotein. One compound, the dual ALK/MET tyrosine kinase inhibitor (TKI), was the first-in-class ALK TKI used in clinics for ALK+ ALCL and has been shown to be effective in refractory/relapsed cases [2]. However, escape mechanisms that allow cancer cells to overcome the effects of crizotinib have emerged [3]. This led to both the development of a new generation of TKI inhibitors as well as a diverse range of combined therapies, in an attempt to preempt relapses and to eradicate the malignant cells [4][5]. In this context, and to improve crizotinib therapy, we investigated the role and regulation of macro-autophagy (hereafter referred to as autophagy) in crizotinib-treated ALK+ ALCL [6][7][8][9][10].
Autophagy is a conserved vesicular pathway that allows cells to sequester and degrade either bulk cytoplasm and/or selective substrates [11]. Such unwanted materials are sealed in double-membrane autophagosomes, and then submitted to degradation by lytic enzymes. These catabolic reactions occur in autophagolysosomes, which are organelles resulting from the fusion between autophagosomes and lysosomes. Being such an essential biological pathway for cell homeostasis and integrity, dysregulations of the autophagic process have been implicated in many diseases, including cancers [12]. In this context, autophagy was shown to prevent tumorigenesis in early stages but favor tumor progression in advanced stages [13]. This two-faced role of autophagy is also observed following cancer therapies. Indeed, depending on the treatments and on the types of cancer, evidence for pro-survival or, conversely, pro-death functions of autophagy has been reported [14][15][16][17].
Over the last few years, our work has focused on the therapeutic modulation of autophagy to improve crizotinib therapy in ALK+ ALCL. Here, we briefly present our previous results, first demonstrating the cytoprotective role of autophagy in bulk and stem-like ALK+ ALCL cells that were submitted to crizotinib single targeted therapy, and we highlight the possible molecular mechanisms underlying this pro-survival function of autophagy. Second, it is showed, on the contrary, pro-death autophagy function in ALK+ ALCL cells submitted to combined therapies (ALK and BCL2 or ALK and RAF1 co-inhibition) and we document as well, from the literature, the proposed molecular mechanisms for tumor cells’ demise in these settings.
2. Pro-Survival Autophagy in Crizotinib-Treated Bulk and Stem-like ALK+ ALCL cells
Evidence for autophagy induction. A body of experiments demonstrated the induction of autophagy in ALK+ ALCL cell lines, following crizotinib treatment. This was assessed by a combination of complementary approaches: increased number of degradative autophagic vacuoles (as detected by electron microscopy), increased LC3-II dot staining (as revealed by immunohistochemistry) and increased expression of autophagy-related genes [6][10]. Moreover, autophagy flux activation was demonstrated by classical LC3 turnover assays, and by using tandem fluorescent probe LC3 expressing ALK+ ALCL cells in both immunofluorescence microscopy and flow cytometry analyses [8][9][10]. Importantly, the autophagy flux was found higher in the stem-like subset of ALK+ ALCL cells, and associated with crizotinib chemo-resistance.
Evidence for pro-survival function of autophagy. It further demonstrated, both in vitro (by performing viability, apoptosis and clonogenic assays) and in vivo (by measuring xenografted tumor growth), that the combination of ALK and autophagy inhibitions led to the re-sensitization of the bulk and stem-like subset of ALK+ ALCL cells to crizotinib thus highlighting the cytoprotective function of autophagy in these settings [6][10].
Possible molecular mechanisms for the cytoprotective autophagy. Autophagy could protect therapeutically challenged tumor cells through (i) the impairment of apoptosis. Indeed, autophagy could sequester and degrade pro-apoptosis proteins [18] (or the upstream transcriptional factors controlling their expression [19]). It could also limit the production of pro-apoptotic reactive oxygen species (ROS) and therefore restrain the oxidative stress-induced apoptotic cell death [20]. Autophagy cytoprotective functions could also rely on (ii) the induction and maintenance of cancer stemness. Indeed, a link between autophagy, cancer stemness and chemoresistance has been described in several cancer types [21] and was correlated, in ALK+ ALCL stem-like subset, with a high protein level of MYC [10]. The downstream effectors of MYC in this particular setting are not known, yet.
3. Autophagy associated with Cell Death in Crizotinib-Treated ALK+ ALCL cells
Definition. The term “Autophagy associated with cell death” encompasses cases of cell death, where autophagy markers and autophagy flux are increased, but where autophagy inhibition does not suppress the death outcome. As detailed below, two of our studies report combined therapies in ALK+ ALCL cells, which result in enhanced autophagy associated with lymphoma cells’ demise.
The combined targeting of ALK and BCL2. We reported that crizotinib treatment resulted in the upregulation of BCL2 mRNA and protein levels. The knockdown of BCL2, concomitantly to crizotinib treatment, resulted in an increased loss in cell viability, associated with enhanced autophagy and apoptosis [22]. In these settings, the blockade of autophagy resulted in a partial rescue in the loss of cell viability. Thus, according to the definition explained above, autophagy appeared to be associated with lymphoma cells’ death.
The combined targeting of ALK and RAF1. We found that RAF1 phosphorylated ULK1 at inhibitory site. Consequently, we found that RAF1 inactivation unleashed the autophagy response, which was associated with increased cell death, including apoptosis [9]. As autophagy inhibition in these settings was not able to rescue cell viability, we proposed that the enhanced autophagy flux was likely to support other cell death modalities, but then became dispensable for the execution of cell death.
Possible molecular mechanisms for pro-death autophagy. In the literature, it has been shown that enhanced autophagy could lead to the death of therapeutically challenged tumor cells through (i) the selective removal of pro-survival substrates [23][24] or anti-apoptosis factors [25]. Autophagy lethal functions could also rely on (ii) autophagosomal membranes, which could serve as scaffold for other cell death modalities, such as apoptosis [26] or necroptosis [27]. Another emerging possibility is related to (iii) the role of autophagy and autophagosomes in antigen presentation[28][29] and in the immune control of tumors [30][31].
4. Conclusion
Autophagy is a fundamental cellular process, controlling cell fate (survival or death) according to the physiological, pathological, and therapeutic context. In cancers, many studies have been conducted to understand how to manipulate the autophagy process to improve treatments. Our work in the field of ALK-targeted therapy in ALK+ ALCL cells demonstrated that “restrained” autophagy (upon crizotinib single treatment) was cytoprotective in bulk and stem-like cells, whereas “enhanced” autophagy (upon combined therapies using crizotinib and additional drugs targeting the autophagy process or one of its regulators) was associated with cell death. Thus, our results indicated that manipulating autophagy could improve the efficiency of crizotinib-targeted therapy.
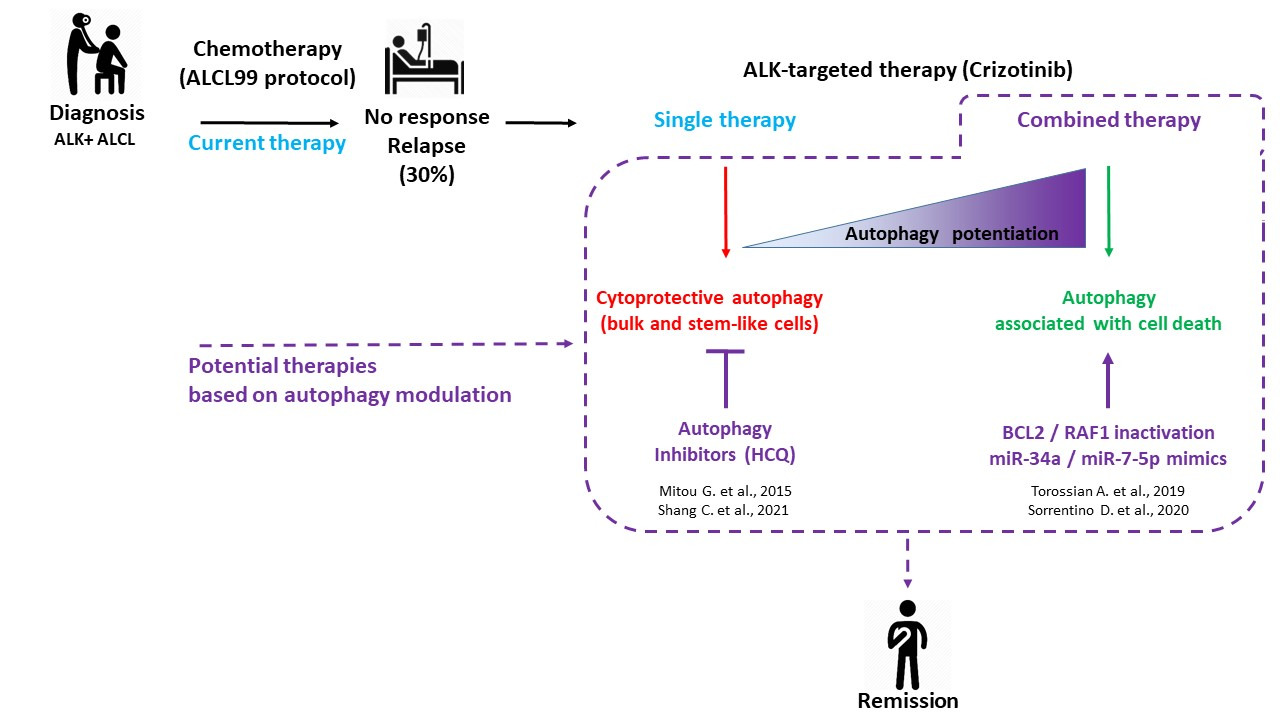
Figure 1. ALK+ ALCL treatments: current and potential therapies based on autophagy modulation. Considering the 30% of ALK+ ALCL patients who are either refractory to the gold standard ALCL99 protocol or who experienced relapses, research efforts were conducted toward the development of targeted and combined therapies. This scheme summarizes our four fundamental studies, showing the potential therapeutic benefit of modulating autophagy to improve the targeted therapy of ALK+ ALCL using crizotinib (dashed lines). Consistent with the known dual role of autophagy (pro-survival or pro-death) according to the therapeutic context, we found that a single treatment of crizotinib led to protective autophagy in bulk and stem-like ALK+ ALCL cells, whereas its combination with inhibitors of other key cellular factors triggered an enhanced autophagy associated with cell death.
In a more global view, it is likely that the assessment of autophagy activity (through genome sequencing and autophagy-related gene expression profiling), both in tumor cells and in their microenvironment, at diagnosis and/or following therapy, could lead to the identification of new molecular biomarkers and actionable biologic pathways, which would potentially improve the clinical management of cancers.
References
- Lowe, E.J.; Gross, T.G. Anaplastic large cell lymphoma in children and adolescents. Pediatr. Hematol. Oncol. 2013, 30, 509–19, doi:10.3109/08880018.2013.805347.
- Gambacorti Passerini, C.; Farina, F.; Stasia, A.; Redaelli, S.; Ceccon, M.; Mologni, L.; Messa, C.; Guerra, L.; Giudici, G.; Sala, E.; et al. Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J Natl Cancer Inst 2014, 106, djt378.
- Sharma, G.G.; Mota, I.; Mologni, L.; Patrucco, E.; Gambacorti-Passerini, C.; Chiarle, R. Tumor Resistance against ALK Targeted Therapy-Where It Comes From and Where It Goes. Cancers (Basel). 2018, 10, 62, doi:10.3390/cancers10030062.
- Mologni, L. Inhibitors of the anaplastic lymphoma kinase. Expert Opin. Investig. Drugs 2012, 21, 985–994, doi:10.1517/13543784.2012.690031.
- Larose, H.; Burke, G.A.A.; Lowe, E.J.; Turner, S.D. From bench to bedside: the past, present and future of therapy for systemic paediatric ALCL, ALK+. Br. J. Haematol. 2019, 185, 1043–1054, doi:10.1111/bjh.15763.
- Mitou, G.; Frentzel, J.; Desquesnes, A.; Le Gonidec, S.; AlSaati, T.; Beau, I.; Lamant, L.; Meggetto, F.; Espinos, E.; Codogno, P.; et al. Targeting autophagy enhances the anti-tumoral action of crizotinib in ALK-positive anaplastic large cell lymphoma. Oncotarget 2015, 6, doi:10.18632/oncotarget.4999.
- Frentzel, J.; Sorrentino, D.; Giuriato, S. Targeting autophagy in ALK-associated cancers. Cancers (Basel). 2017, 9, 161, doi:10.3390/cancers9120161.
- Torossian, A.; Broin, N.; Frentzel, J.; Daugrois, C.; Gandarillas, S.; Al Saati, T.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Blockade of crizotinib-induced BCL2 elevation in ALK-positive anaplastic large cell lymphoma triggers autophagy associated with cell death. Haematologica 2019, 104, doi:10.3324/haematol.2017.181966.
- Sorrentino, D.; Frentzel, J.; Mitou, G.; Blasco, R.B.; Torossian, A.; Hoareau-Aveilla, C.; Pighi, C.; Farcé, M.; Meggetto, F.; Manenti, S.; et al. High levels of mir-7-5p potentiate crizotinib-induced cytokilling and autophagic flux by targeting raf1 in npm-alk positive lymphoma cells. Cancers (Basel). 2020, 12, 1–22, doi:10.3390/cancers12102951.
- Shang, C.; Hassan, B.; Haque, M.; Song, Y.; Li, J.; Liu, D.; Lipke, E.; Chen, W.; Giuriato, S.; Lai, R. Crizotinib resistance mediated by autophagy is higher in the stem-like cell subset in ALK-positive anaplastic large cell lymphoma, and this effect is MYC-dependent. Cancers (Basel). 2021, 13, 1–16, doi:10.3390/cancers13020181.
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 521–527, doi:10.1038/s41556-018-0092-5.
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 69–79, doi:10.1038/cr.2013.161.
- Galluzzi, L.; Pietrocola, F.; Pedro, J.M.B.; Amaravadi, R.K.; Baehrecke, E.H.; Cecconi, F.; Codogno, P.; Debnath, J.; Gewirtz, D.A.; Karantza, V.; et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015, 34, e201490784, doi:10.15252/embj.201490784.
- Rosenfeldt, M.T.; Ryan, K.M. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med 2009, 11, e36.
- Joffre, C.; Djavaheri-Mergny, M.; Pattingre, S.; Giuriato, S. The yin and the yang of autophagy in cancer cells. Medecine/Sciences 2017, 33, 328–334, doi:10.1051/medsci/20173303021.
- Djavaheri-Mergny, M.; Giuriato, S.; Tschan, M.P.; Humbert, M. Therapeutic Modulation of Autophagy in Leukaemia and Lymphoma. Cells 2019, 8, 103, doi:10.3390/cells8020103.
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137, doi:10.1016/j.ejps.2019.04.011.
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Autophagic degradation of active caspase-8: A crosstalk mechanism between autophagy and apoptosis. Autophagy 2010, 6, 891–900, doi:10.4161/auto.6.7.13038.
- Fitzwalter, B.E.; Towers, C.G.; Sullivan, K.D.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy Inhibition Mediates Apoptosis Sensitization in Cancer Therapy by Relieving FOXO3a Turnover. Dev. Cell 2018, 44, 555-565.e3, doi:10.1016/j.devcel.2018.02.014.
- Shi, Y.; Tang, B.; Yu, P.W.; Tang, B.; Hao, Y.X.; Lei, X.; Luo, H.X.; Zeng, D.Z. Autophagy Protects against Oxaliplatin-Induced Cell Death via ER Stress and ROS in Caco-2 Cells. PLoS One 2012, 7, e51076, doi:10.1371/journal.pone.0051076.
- Mandhair, H.K.; Arambasic, M.; Novak, U.; Radpour, R. Molecular modulation of autophagy: New venture to target resistant cancer stem cells. World J. Stem Cells 2020, 12, 303–322, doi:10.4252/wjsc.v12.i5.303.
- Torossian, A.; Broin, N.; Frentzel, J.; Daugrois, C.; Gandarillas, S.; Al Saati, T.; Lamant, L.; Brousset, P.; Giuriato, S.; Espinos, E. Blockade of crizotinib-induced BCL2 elevation in ALK-positive anaplastic large cell lymphoma triggers autophagy associated with cell death. Haematologica 2019, 104, 1428–1439, doi:10.3324/haematol.2017.181966.
- Goussetis, D.J.; Gounaris, E.; Wu, E.J.; Vakana, E.; Sharma, B.; Bogyo, M.; Altman, J.K.; Platanias, L.C. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood 2012, 120, 3555–3562, doi:10.1182/blood-2012-01-402578blood-2012-01-402578 [pii].
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.H.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 4952–4957, doi:10.1073/pnas.0511288103.
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.H.; Thorburn, A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54, doi:10.1038/ncb2886.
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.-D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–68, doi:10.1074/jbc.M111.309104.
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173, doi:10.1038/cdd.2013.45.
- Li, B.; Lei, Z.; Lichty, B.D.; Li, D.; Zhang, G.M.; Feng, Z.H.; Wan, Y.; Huang, B. Autophagy facilitates major histocompatibility complex class I expression induced by IFN-γ in B16 melanoma cells. Cancer Immunol. Immunother. 2010, 59, 313–321, doi:10.1007/s00262-009-0752-1.
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92, doi:10.1016/j.immuni.2006.10.018.
- de Souza, A.S.C.; Gonçalves, L.B.; Lepique, A.P.; de Araujo-Souza, P.S. The Role of Autophagy in Tumor Immunology—Complex Mechanisms That May Be Explored Therapeutically. Front. Oncol. 2020.
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021.

