 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Daniel Romaus-Sanjurjo | + 3259 word(s) | 3259 | 2021-12-08 03:03:49 | | | |
| 2 | Rita Xu | Meta information modification | 3259 | 2021-12-10 06:41:06 | | |
Video Upload Options
In 1959, E. G. Gray described two different types of synapses in the brain for the first time: symmetric and asymmetric. Later on, symmetric synapses were associated with inhibitory terminals, and asymmetric synapses to excitatory signaling. The balance between these two systems is critical to maintain a correct brain function. Likewise, the modulation of both types of synapses is also important to maintain a healthy equilibrium. Cerebral circuitry responds differently depending on the type of damage and the timeline of the injury. For example, promoting symmetric signaling following ischemic damage is beneficial only during the acute phase; afterwards, it further increases the initial damage. Synapses can be also altered by players not directly related to them; the chronic and long-term neurodegeneration mediated by tau proteins primarily targets asymmetric synapses by decreasing neuronal plasticity and functionality. Dopamine represents the main modulating system within the central nervous system. Indeed, the death of midbrain dopaminergic neurons impairs locomotion, underlying the devastating Parkinson’s disease.
1. Introduction
2. Ischemic Stroke
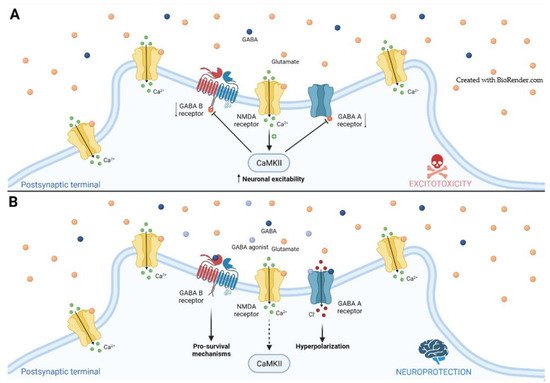
2.1. GABA Receptors
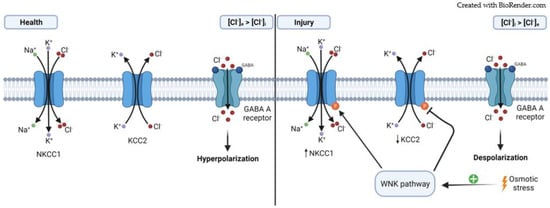
2.2. Cation–Chloride Cotransporters
References
- Gray, E.G. Axo-somatic and axo-dendritic synapses of the cerebral cortex: An electron microscope study. J. Anat. 1959, 93, 420–433.
- Colonnier, M. Synaptic patterns on different cell types in the different laminae of the cat visual cortex. An electron microscope study. Brain Res. 1968, 9, 268–287.
- Klemann, C.J.; Roubos, E.W. The gray area between synapse structure and function-Gray’s synapse types I and II revisited. Synapse 2011, 65, 1222–1230.
- Siekevitz, P. The postsynaptic density: A possible role in long-lasting effects in the central nervous system. Proc. Natl. Acad. Sci. USA 1985, 82, 3494–3498.
- Parato, J.; Bartolini, F. The microtubule cytoskeleton at the synapse. Neurosci. Lett. 2021, 753, 135850.
- Moraes, B.J.; Coelho, P.; Fão, L.; Ferreira, I.L.; Rego, A.C. Modified Glutamatergic Postsynapse in Neurodegenerative Disorders. Neuroscience 2021, 454, 116–139.
- Smart, T.G.; Paoletti, P. Synaptic neurotransmitter-gated receptors. Cold Spring Harb. Perspect. Biol. 2012, 4, a009662.
- Sheng, M.; Kim, E. The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 2011, 3, a005678.
- Rodzli, N.A.; Lockhart-Cairns, M.P.; Levy, C.W.; Chipperfield, J.; Bird, L.; Baldock, C.; Prince, S.M. The Dual PDZ Domain from Postsynaptic Density Protein 95 Forms a Scaffold with Peptide Ligand. Biophys J. 2020, 119, 667–689.
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781.
- Luscher, B.; Fuchs, T.; Kilpatrick, C.L. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron 2011, 70, 385–409.
- Tyagarajan, S.K.; Fritschy, J.M. Gephyrin: A master regulator of neuronal function? Nat. Rev. Neurosci. 2014, 15, 141–156.
- Pizzarelli, R.; Griguoli, M.; Zacchi, P.; Petrini, E.M.; Barberis, A.; Cattaneo, A.; Cherubini, E. Tuning GABAergic Inhibition: Gephyrin Molecular Organization and Functions. Neuroscience 2020, 439, 125–136.
- Abraham, W.C.; Jones, O.D.; Glanzman, D.L. Is plasticity of synapses the mechanism of long-term memory storage? NPJ Sci. Learn. 2019, 4, 9.
- Madadi Asl, M.; Vahabie, A.H.; Valizadeh, A. Dopaminergic Modulation of Synaptic Plasticity, Its Role in Neuropsychiatric Disorders, and Its Computational Modeling. Basic Clin. Neurosci. 2019, 10, 1–12.
- Malenka, R.C. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 1994, 78, 535–538.
- Bloodgood, B.L.; Giessel, A.J.; Sabatini, B.L. Biphasic synaptic Ca influx arising from compartmentalized electrical signals in dendritic spines. PLoS Biol. 2009, 7, e1000190.
- Castillo, P.E.; Chiu, C.Q.; Carroll, R.C. Long-term plasticity at inhibitory synapses. Curr. Opin. Neurobiol. 2011, 21, 328–338.
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710.
- Nadim, F.; Bucher, D. Neuromodulation of neurons and synapses. Curr. Opin. Neurobiol. 2014, 29, 48–56.
- Chinta, S.J.; Andersen, J.K. Dopaminergic neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946.
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50.
- Pedrosa, V.; Clopath, C. The Role of Neuromodulators in Cortical Plasticity. A Computational Perspective. Front Synaptic. Neurosci. 2017, 8, 38.
- Béjot, Y.; Bailly, H.; Durier, J.; Giroud, M. Epidemiology of stroke in Europe and trends for the 21st century. Presse Med. 2016, 45, e391–e398.
- Hay, B.; Yi, S.; Patel, P. Cerebral Ischemia. In Gupta and Gelb’s Essentials of Neuroanesthesia and Neurointensive Care, 2nd ed.; Gupta, A., Gelb, A., Duane, D., Adapa, R., Eds.; Cambridge University Press: Cambridge, UK, 2018; pp. 39–47.
- Martín-Aragón Baudel, M.A.; Poole, A.V.; Darlison, M.G. Chloride co-transporters as possible therapeutic targets for stroke. J. Neurochem. 2017, 140, 195–209.
- Rahman, A.A.; Amruta, N.; Pinteaux, E.; Bix, G.J. Neurogenesis After Stroke: A Therapeutic Perspective. Transl. Stroke Res. 2021, 12, 1–14.
- Clarkson, A.N.; Huang, B.S.; Macisaac, S.E.; Mody, I.; Carmichael, S.T. Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke. Nature 2010, 468, 305–309.
- Hiu, T.; Farzampour, Z.; Paz, J.T.; Wang, E.H.; Badgely, C.; Olson, A.; Micheva, K.D.; Wang, G.; Lemmens, R.; Tran, K.V.; et al. Enhanced phasic GABA inhibition during the repair phase of stroke: A novel therapeutic target. Brain 2016, 139, 468–480.
- Mele, M.; Costa, R.O.; Duarte, C.B. Alterations in GABAA-Receptor Trafficking and Synaptic Dysfunction in Brain Disorders. Front Cell Neurosci. 2019, 13, 77.
- Tanaka, E.; Ogawa, Y.; Fujii, R.; Shimonaka, T.; Sato, Y.; Hamazaki, T.; Nagamura-Inoue, T.; Shintaku, H.; Tsuji, M. Metabolomic analysis and mass spectrometry imaging after neonatal stroke and cell therapies in mouse brains. Sci. Rep. 2020, 10, 21881.
- Mele, M.; Leal, G.; Duarte, C.B. Role of GABAA R trafficking in the plasticity of inhibitory synapses. J. Neurochem. 2016, 139, 997–1018.
- Carmichael, S.T. Brain excitability in stroke: The yin and yang of stroke progression. Arch. Neurol. 2012, 69, 161–167.
- Alia, C.; Spalletti, C.; Lai, S.; Panarese, A.; Micera, S.; Caleo, M. Reducing GABAA-mediated inhibition improves forelimb motor function after focal cortical stroke in mice. Sci. Rep. 2016, 6, 37823.
- Hagemann, G.; Redecker, C.; Neumann-Haefelin, T.; Freund, H.J.; Witte, O.W. Increased long-term potentiation in the surround of experimentally induced focal cortical infarction. Ann. Neurol. 1998, 44, 255–258.
- Cramer, S.C. Repairing the human brain after stroke: I. Mechanisms of spontaneous recovery. Ann. Neurol. 2008, 63, 272–287.
- Wang, Y.C.; Dzyubenko, E.; Sanchez-Mendoza, E.H.; Sardari, M.; Silva de Carvalho, T.; Doeppner, T.R.; Kaltwasser, B.; Machado, P.; Kleinschnitz, C.; Bassetti, C.L.; et al. Postacute Delivery of GABAA α5 Antagonist Promotes Postischemic Neurological Recovery and Peri-infarct Brain Remodeling. Stroke 2018, 49, 2495–2503.
- van Nieuwenhuijzen, P.S.; Parker, K.; Liao, V.; Houlton, J.; Kim, H.L.; Johnston, G.A.R.; Hanrahan, J.R.; Chebib, M.; Clarkson, A.N. Targeting GABAC Receptors Improves Post-Stroke Motor Recovery. Brain Sci. 2021, 11, 315.
- Chalifoux, J.R.; Carter, A.G. GABAB receptor modulation of synaptic function. Curr. Opin. Neurobiol. 2011, 21, 339–344.
- Mele, M.; Ribeiro, L.; Inácio, A.R.; Wieloch, T.; Duarte, C.B. GABA(A) receptor dephosphorylation followed by internalization is coupled to neuronal death in in vitro ischemia. Neurobiol. Dis. 2014, 65, 220–232.
- Hausrat, T.J.; Muhia, M.; Gerrow, K.; Thomas, P.; Hirdes, W.; Tsukita, S.; Heisler, F.F.; Herich, L.; Dubroqua, S.; Breiden, P.; et al. Radixin regulates synaptic GABAA receptor density and is essential for reversal learning and short-term memory. Nat. Commun. 2015, 6, 6872.
- Costa, J.T.; Mele, M.; Baptista, M.S.; Gomes, J.R.; Ruscher, K.; Nobre, R.J.; de Almeida, L.P.; Wieloch, T.; Duarte, C.B. Gephyrin Cleavage in In Vitro Brain Ischemia Decreases GABAA Receptor Clustering and Contributes to Neuronal Death. Mol. Neurobiol. 2016, 53, 3513–3527.
- Benke, D.; Balakrishnan, K.; Zemoura, K. Regulation of cell surface GABA(B) receptors: Contribution to synaptic plasticity in neurological diseases. Adv. Pharm. 2015, 73, 41–70.
- Huang, L.; Li, Q.; Wen, R.; Yu, Z.; Li, N.; Ma, L.; Feng, W. Rho-kinase inhibitor prevents acute injury against transient focal cerebral ischemia by enhancing the expression and function of GABA receptors in rats. Eur. J. Pharm. 2017, 797, 134–142.
- Zagrean, A.M.; Grigoras, I.F.; Iesanu, M.I.; Ionescu, R.B.; Chitimus, D.M.; Haret, R.M.; Ianosi, B.; Ceanga, M.; Zagrean, L. Neuronal Transmembrane Chloride Transport Has a Time-Dependent Influence on Survival of Hippocampal Cultures to Oxygen-Glucose Deprivation. Brain. Sci. 2019, 9, 360.
- Zemoura, K.; Balakrishnan, K.; Grampp, T.; Benke, D. Ca2+/Calmodulin-Dependent Protein Kinase II (CaMKII) β-Dependent Phosphorylation of GABAB1 Triggers Lysosomal Degradation of GABAB Receptors via Mind Bomb-2 (MIB2)-Mediated Lys-63-Linked Ubiquitination. Mol. Neurobiol. 2019, 56, 1293–1309.
- García-Berrocoso, T.; Llombart, V.; Colàs-Campàs, L.; Hainard, A.; Licker, V.; Penalba, A.; Ramiro, L.; Simats, A.; Bustamante, A.; Martínez-Saez, E.; et al. Single Cell Immuno-Laser Microdissection Coupled to Label-Free Proteomics to Reveal the Proteotypes of Human Brain Cells After Ischemia. Mol. Cell. Proteom. 2018, 17, 175–189.
- Ramiro, L.; García-Berrocoso, T.; Briansó, F.; Goicoechea, L.; Simats, A.; Llombart, V.; Gonzalo, R.; Hainard, A.; Martínez-Saez, E.; Canals, F.; et al. Integrative Multi-omics Analysis to Characterize Human Brain Ischemia. Mol. Neurobiol. 2021, 58, 4107–4121.
- Costa, C.; Leone, G.; Saulle, E.; Pisani, F.; Bernardi, G.; Calabresi, P. Coactivation of GABA(A) and GABA(B) receptor results in neuroprotection during in vitro ischemia. Stroke 2004, 35, 596–600.
- Yuan, Y.J.; Ye, Z.; Yu, H.; Chen, Y.; Wang, Y.W.; Zhao, J.H.; Sun, J.F.; Xu, L.M. Shrm4 contributes to autophagy inhibition and neuroprotection following ischemic stroke by mediating GABAB receptor activation. FASEB J. 2020, 34, 15837–15848.
- Lyden, P.D.; Hedges, B. Protective effect of synaptic inhibition during cerebral ischemia in rats and rabbits. Stroke 1992, 23, 1463–1470.
- Shames, J.L.; Ring, H. Transient reversal of anoxic brain injury-related minimally conscious state after zolpidem administration: A case report. Arch. Phys. Med. Rehabil. 2008, 89, 386–388.
- Hall, S.D.; Yamawaki, N.; Fisher, A.E.; Clauss, R.P.; Woodhall, G.L.; Stanford, I.M. GABA(A) alpha-1 subunit mediated desynchronization of elevated low frequency oscillations alleviates specific dysfunction in stroke--a case report. Clin. Neurophysiol. 2010, 121, 549–555.
- Hummel, F.C.; Cohen, L.G. Non-invasive brain stimulation: A new strategy to improve neurorehabilitation after stroke? Lancet Neurol. 2006, 5, 708–712.
- Kokinovic, B.; Medini, P. Loss of GABAB -mediated interhemispheric synaptic inhibition in stroke periphery. J. Physiol. 2018, 596, 1949–1964.
- Cirillo, J.; Mooney, R.A.; Ackerley, S.J.; Barber, P.A.; Borges, V.M.; Clarkson, A.N.; Mangold, C.; Ren, A.; Smith, M.C.; Stinear, C.M.; et al. Neurochemical balance and inhibition at the subacute stage after stroke. J. Neurophysiol. 2020, 123, 1775–1790.
- Schulte, J.T.; Wierenga, C.J.; Bruining, H. Chloride transporters and GABA polarity in developmental, neurological and psychiatric conditions. Neurosci. Biobehav. Rev. 2018, 90, 260–271.
- Romaus-Sanjurjo, D.; Rodicio, M.C.; Barreiro-Iglesias, A. Gamma-aminobutyric acid (GABA) promotes recovery from spinal cord injury in lampreys: Role of GABA receptors and perspective on the translation to mammals. Neural Regen. Res. 2019, 14, 1695–1696.
- Ben-Ari, Y. NKCC1 Chloride Importer Antagonists Attenuate Many Neurological and Psychiatric Disorders. Trends Neurosci. 2017, 40, 536–554.
- Wang, G.; Huang, H.; He, Y.; Ruan, L.; Huang, J. Bumetanide protects focal cerebral ischemia-reperfusion injury in rat. Int. J. Clin. Exp. Pathol. 2014, 7, 1487–1494.
- Zhang, J.; Gao, G.; Begum, G.; Wang, J.; Khanna, A.R.; Shmukler, B.E.; Daubner, G.M.; de Los Heros, P.; Davies, P.; Varghese, J.; et al. Functional kinomics establishes a critical node of volume-sensitive cation-Cl− cotransporter regulation in the mammalian brain. Sci. Rep. 2016, 6, 35986.
- Josiah, S.S.; Meor Azlan, N.F.; Zhang, J. Targeting the WNK-SPAK/OSR1 Pathway and Cation-Chloride Cotransporters for the Therapy of Stroke. Int. J. Mol. Sci. 2021, 22, 1232.
- Yan, Y.; Dempsey, R.J.; Flemmer, A.; Forbush, B.; Sun, D. Inhibition of Na(+)-K(+)-Cl(-) cotransporter during focal cerebral ischemia decreases edema and neuronal damage. Brain Res. 2003, 961, 22–31.
- Jaenisch, N.; Witte, O.W.; Frahm, C. Downregulation of potassium chloride cotransporter KCC2 after transient focal cerebral ischemia. Stroke 2010, 41, e151–e159.
- Begum, G.; Yuan, H.; Kahle, K.T.; Li, L.; Wang, S.; Shi, Y.; Shmukler, B.E.; Yang, S.S.; Lin, S.H.; Alper, S.L.; et al. Inhibition of WNK3 Kinase Signaling Reduces Brain Damage and Accelerates Neurological Recovery After Stroke. Stroke 2015, 46, 1956–1965.
- Mu, X.P.; Wang, H.B.; Cheng, X.; Yang, L.; Sun, X.Y.; Qu, H.L.; Zhao, S.S.; Zhou, Z.K.; Liu, T.T.; Xiao, T.; et al. Inhibition of Nkcc1 promotes axonal growth and motor recovery in ischemic rats. Neuroscience 2017, 365, 83–93.
- Glykys, J.; Dzhala, V.; Egawa, K.; Kahle, K.T.; Delpire, E.; Staley, K. Chloride Dysregulation, Seizures, and Cerebral Edema: A Relationship with Therapeutic Potential. Trends Neurosci. 2017, 40, 276–294.
- Xu, W.; Mu, X.; Wang, H.; Song, C.; Ma, W.; Jolkkonen, J.; Zhao, C. Chloride Co-transporter NKCC1 Inhibitor Bumetanide Enhances Neurogenesis and Behavioral Recovery in Rats After Experimental Stroke. Mol. Neurobiol. 2017, 54, 2406–2414.
- Huang, H.; Song, S.; Banerjee, S.; Jiang, T.; Zhang, J.; Kahle, K.T.; Sun, D.; Zhang, Z. The WNK-SPAK/OSR1 Kinases and the Cation-Chloride Cotransporters as Therapeutic Targets for Neurological Diseases. Aging Dis. 2019, 10, 626–636.
- Bhuiyan, M.I.H.; Song, S.; Yuan, H.; Begum, G.; Kofler, J.; Kahle, K.T.; Yang, S.S.; Lin, S.H.; Alper, S.L.; Subramanya, A.R.; et al. WNK-Cab39-NKCC1 signaling increases the susceptibility to ischemic brain damage in hypertensive rats. J. Cereb. Blood Flow Metab. 2017, 37, 2780–2794.
- Shekarabi, M.; Zhang, J.; Khanna, A.R.; Ellison, D.H.; Delpire, E.; Kahle, K.T. WNK Kinase Signaling in Ion Homeostasis and Human Disease. Cell Metab. 2017, 25, 285–299.
- Zhang, J.; Bhuiyan, M.I.H.; Zhang, T.; Karimy, J.K.; Wu, Z.; Fiesler, V.M.; Zhang, J.; Huang, H.; Hasan, M.N.; Skrzypiec, A.E.; et al. Modulation of brain cation-Cl− cotransport via the SPAK kinase inhibitor ZT-1a. Nat. Commun. 2020, 11, 78.
- Pin-Barre, C.; Constans, A.; Brisswalter, J.; Pellegrino, C.; Laurin, J. Effects of High-Versus Moderate-Intensity Training on Neuroplasticity and Functional Recovery After Focal Ischemia. Stroke 2017, 48, 2855–2864.
- Khirug, S.; Soni, S.; Saez Garcia, M.; Tessier, M.; Zhou, L.; Kulesskaya, N.; Rauvala, H.; Lindholm, D.; Ludwig, A.; Molinari, F.; et al. Protective Role of Low Ethanol Administration Following Ischemic Stroke via Recovery of KCC2 and p75NTR Expression. Mol. Neurobiol. 2021, 58, 1145–1161.

