In 1959, E. G. Gray described two different types of synapses in the brain for the first time: symmetric and asymmetric. Later on, symmetric synapses were associated with inhibitory terminals, and asymmetric synapses to excitatory signaling. The balance between these two systems is critical to maintain a correct brain function. Likewise, the modulation of both types of synapses is also important to maintain a healthy equilibrium. Cerebral circuitry responds differently depending on the type of damage and the timeline of the injury. For example, promoting symmetric signaling following ischemic damage is beneficial only during the acute phase; afterwards, it further increases the initial damage. Synapses can be also altered by players not directly related to them; the chronic and long-term neurodegeneration mediated by tau proteins primarily targets asymmetric synapses by decreasing neuronal plasticity and functionality. Dopamine represents the main modulating system within the central nervous system. Indeed, the death of midbrain dopaminergic neurons impairs locomotion, underlying the devastating Parkinson’s disease.
1. Introduction
At the end of the 1950′s, E. G. Gray used electron microscopy to define two different types of synapses in the central nervous system (CNS): asymmetric and symmetric synapses
[1]. Based on his achievements, asymmetric (or type I) synapses are defined by a postsynaptic density (PSD), thicker than the presynaptic fraction, whereas symmetric (or type II) synapses present a PSD similar in width to the presynaptic membrane. Subsequently, asymmetric and symmetric synapses were correlated to excitatory or inhibitory signaling, respectively
[2]. Although controversial
[3], nowadays this terminology is still being used to identify excitatory and inhibitory synapses along CNS.
As mentioned, the PSD is a high-density fraction in the postsynaptic membrane with different roles such as mediating the apposition of pre- and post-synaptic membranes, clustering postsynaptic receptors or coupling the activation of these receptors to cellular signaling
[4][5][6][4,5,6]. The PSD in asymmetric synapses is composed of membrane proteins (e.g., α-amino-3-hidroxi-5-metilo-4-isoxazolpropionic receptor [AMPAR], N-methyl-D-aspartate receptor [NMDAR], metabotropic receptors, ion channels and adhesion molecules), scaffold proteins (such as the postsynaptic density protein 95 [PSD-95]), and signaling proteins
[6][7][6,7]. PSD-95 is the most abundant scaffold protein in postsynapses, where it plays a crucial role in organization by interacting with adhesion molecules, glutamate receptors and signaling proteins through its PDZ domain
[8][9][8,9]. Accordingly, high levels of PSD-95 are correlated with larger PSDs and enhanced synaptic strength
[10]. In contrast, symmetric synapses display a different composition in their PSD, where gamma-aminobutyric acid (GABA) A (GABA
A, ionotropic) and GABA B (GABA
B, metabotropic) receptors are responsible for mediating inhibitory responses. Interestingly, the number of GABA
A receptors at the membrane usually determines the strength of the inhibitory synaptic signaling
[11]. Similarly to PSD-95, gephyrin plays an important role in the structure of the inhibitory PSD by clustering GABA receptors and acting as a scaffold protein
[12][13][12,13]. Both asymmetric and symmetric PSDs are not fixed but constantly changing, reflecting the high plasticity presents in this network. The strength of these synapses can be modified in a bidirectional way by mechanisms such as long-term potentiation (LTP) or long-term depression (LTD), among others
[14]. Likewise, modulatory neurotransmitters can also influence and regulate synaptic transmission
[15].
LTP and LTD are well-known forms of synaptic plasticity. Most of our knowledge about LTP/LTD came from reports of asymmetric synapses, where NMDA-mediated LTP/LTD is the most studied
[14]. In excitatory synapses, LTP is induced only when both pre- and post-synaptic neurons are active, and it is mandatory that the postsynaptic neuron is already depolarized at the moment glutamate binds to NMDARs. This is important because it is needed to reach the highest calcium influx to activate intracellular signaling pathways underlying these synaptic modifications
[16]. Contrary to LTP, LTD is generally induced by repeated activation of the presynaptic neuron without postsynaptic activity, that leads to a smaller NMDA-mediated calcium influx and synaptic endocytosis of AMPARs
[16][17][16,17]. Regarding inhibitory transmission, LTP/LTD mechanisms are also present in inhibitory synapses throughout the brain
[18]. Inhibitory LTP or LTD needs the presence of glutamatergic synapses, and therefore, the activation of corresponding glutamate receptors to trigger the underlying cellular mechanisms
[19].
Neuromodulators are compounds that modify synaptic transmission by regulating the excitability of both pre- and post-synaptic neurons and the response of receptors to neurotransmitters
[20]. Within neuromodulators, dopamine (DA) is one of the most studied because its functions are of such importance that deficits in dopaminergic (DAergic) signaling lead to neurological disorders
[15]. Midbrain DAergic neurons represent the main source of DA in the CNS, the
substantia nigra pars compacta (SNc) and the ventral tegmental area being two important centers providing significant amount of DA to the basal ganglia (BG) and forebrain
[21]. Through the activation of metabotropic receptors (D1-D5), DA can modify the excitability of neurons by regulating the voltage- or ligand-gated channels
[15], as well as regulating the function and trafficking of GABA receptors, NMDARs, and AMPARs
[22]. In this way, DA is able to affect different synaptic dynamics
[23].
2. Ischemic Stroke
Stroke is becoming one of the most common causes of death in developed countries, representing the main cause of long-term disability due to the limited capacity of human brain to repair. Ischemic stroke, the occlusion of a blood vessel leading to a lack of blood flux, has fatal consequences even in short-term blockages and it represents the 85% of total cases in Europe
[24][25][24,25]. Following the insult, two large areas can be distinguished: the ischemic core, necrotic tissue with irreparable damage; and the peri-infarct, or penumbra, an area containing hypoperfused tissue that is still viable for several hours and can be salvaged by restoration of the blood flow. Over the next few hours to days, this peri-infarct tissue undergoes secondary damage by the activation of the ischemic cascade which eventually leads to neuronal death. The response to the damage varies depending on which cerebral area is affected, the cortex and hippocampus arising as two of the most susceptible areas
[26][27][26,27]. The timeline of neuronal death differs among these two areas, with cortical neurons displaying a quick death in comparison with hippocampal neurons that show a delayed death occurring 3–5 days following the insult
[26]. This exposes the complexity of neuronal connections since every cortical microcircuit responds differently after damage, and the outcome following treatment may not be the same throughout the different cortical layers
[28][29][28,29].
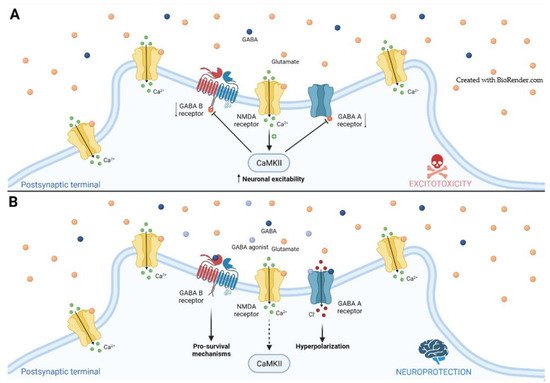
Two different phases can be distinguished from the onset of an ischemic insult, and each one shows how the imbalance between excitatory and inhibitory signaling can negatively affect neuronal/functional outcome
[30]. During the acute phase, under a hypoxic environment, there is a massive presynaptic release of glutamate that overactivates postsynaptic NMDARs. This leads to the entry of large amounts of Ca
2+ during the first minutes to hours, which stimulates a variety of cellular processes that ultimately produce irreparable neuronal damage and cell death (
Figure 1)
[25][30][25,30]. Recently, Tanaka et al.
[31] reported increased levels of glutamate by using MALDI mass spectrometry imaging in the peri-infarct area of a mouse model. In addition to this, the astrogial-mediated reuptake of glutamate is reduced following injury, further increasing extracellular levels of glutamate
[26]. In such a situation, the enhancement of GABA signaling counterbalances the excitatory inputs promoting neuroprotection (
Figure 1)
[32]. Conversely, during the post-acute/chronic phase, GABA signaling is highly increased and limits neural repair by decreasing neuronal excitability and impairing LTP
[33][34][33,34]. This occurs simultaneously with a rearrangement of cortical networks underlying neuronal plasticity by enhancing the ability to induce LTP throughout prolonged excitatory signaling during the first week post stroke
[33][35][36][33,35,36]. Therefore, treatments blocking GABA signaling during this phase may represent promising therapies to help in the recovery of patients following stroke
[28][34][37][38][28,34,37,38].
Figure 1. Glutamate-mediated excitotoxicity in ischemic stroke. (A) Following ischemic injury, the massive release of glutamate (orange spheres) leads to the entry of large amount of Ca2+, increasing neuronal excitability and activating the Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII phosphorylates both GABAA and GABAB receptors, decreasing their availability; (B) The potentiation of symmetric signaling through agonists (light-blue spheres) of either GABAA or GABAB receptor, or GABA itself (dark-blue spheres), counteracts the glutamate-mediated excitotoxicity by hyperpolarizing the neuron and activating pro-survival second messengers that altogether leads to neuroprotection.
Overall, avoiding the transformation of the penumbra into infarcted tissue is a key target to overcome neuronal damage, and it may improve the outcome of patients after stroke. Besides, it seems pivotal to understand how and when the switch from acute to post-acute/chronic phase occurs in humans in order to tackle the distinct cellular mechanisms underlying neuronal damage over time. Achievements in this field will allow the translation from animal models to human.
2.1. GABA Receptors
GABA signaling through the GABA
A receptor is more relevant than the same mediated by GABA
B receptors in the pathophysiology of stroke. Therefore, we will focus primarily on GABA
A receptors, only citing the most relevant information regarding GABA
B receptors.
The different subunits forming ionotropic GABA
A receptors determine the properties and location of receptors. These changes in subunit composition are responsible for the synaptic and extrasynaptic location of GABA
A receptors, which mediate phasic (synaptic) and tonic (extrasynaptic) inhibition, respectively
[32]. During phasic inhibition, GABA released from presynaptic terminals reaches the postsynaptic membrane where it binds to GABA
A receptors and triggers an inward chloride current, leading to the hyperpolarization of the neuron. This cellular mechanism represents a transient response defined by a rapid desensitization of the synaptic GABA
A receptors and the removal of extrasynaptic GABA by GABA transporters (GATs). On the other hand, tonic inhibition mediates a continuously inhibitory current controlling the neuronal membrane potential and thus its fire potential. Such GABAergic signaling is triggered when extrasynaptic GABA
A receptors with high affinity and slow desensitization for GABA respond to either ambient GABA levels outside the synapse or synaptic spillover of GABA. Regarding metabotropic GABA
B receptors, they are the mainly regulators of presynaptic glutamate release in excitatory neurons; they also control the activity of postsynaptic glutamate receptors
[39].
In the GABA
A receptors, trafficking to and from the plasma membrane only occurs at the extrasynaptic space, lateral diffusion being the main mechanism controlling their synaptic pool, and therefore the strength of symmetric signaling
[32]. Based on their location, the clustering of GABA
A receptors is modulated by gephyrin (synaptic site) or radixin (extrasynaptic site), and both scaffold proteins are positively regulated by phosphorylation, strengthen the clustering at the membrane
[32][40][41][32,40,41]. Mele and colleagues
[40] suggested that the dephosphorylation of α1 subunit-containing GABA
A receptors is directly involved in their internalization, likely by losing the link with gephyrin, following in vitro ischemic damage. Likewise, it has been proposed that the calcium-mediated activation of calpain leads to the cleavage of the gephyrin lattice and subsequent reduction in the synaptic clustering of GABA
A receptors in hippocampal neurons from rats under in vitro excitotoxic conditions
[42]. Hence, ischemic conditions lead to decreased levels of phosphorylated GABA
A receptors, as well as GABA
B receptors, suggesting that this is the reason underlying the ischemia-induced endocytosis of receptors. Moreover, this decrease could also explain why GABA
B receptors cannot counteract the glutamate-mediated overexcitation
[43][44][43,44].
Immediately after an ischemic event, large amounts of glutamate contribute to a strong activation of NMDARs that downregulates the expression of both GABA
A and GABA
B receptors through a phosphorylation process activated by high levels of Ca
2+ (
Figure 1)
[32][43][44][45][46][32,43,44,45,46]. Accordingly, phasic GABA signaling is reduced in the first weeks after stroke
[28][40][28,40]. This situation further increases neuronal depolarization and subsequent cellular damage. Recently, two proteomic studies have revealed increased levels of the GABA aminotransferase GABT, as well as reduced levels of GABA receptors and the excitatory amino acid transporter EAA2, in the infarct core area from postmortem tissue samples of stroke patients
[47][48][47,48]. These results validate results from animal models by showing overall decreased GABAergic signaling (elevated catabolism of GABA and reduced GABA receptors) and increased glutamatergic signaling (reduced removal from synaptic cleft by EAA2). That is why the enhancement of GABA signaling at this point can exert a neuroprotective role by decreasing the cellular excitability (
Figure 1). Indeed, an early study by Costa and coworkers
[49] revealed that the coactivation of both GABA
A and GABA
B receptors promoted neuroprotection in an in vitro model of ischemic stroke. Similarly, the activation of either GABA
A or GABA
B receptors separately also has pro-survival outcomes. Several studies have reported that the remaining GABA
B receptors can be activated between days 1–3 post stroke and this promotes neuroprotection
[42][50][42,50]. Since the 1990′s, the neuroprotective role of enhancing phasic GABA signaling at the acute phase has been studied throughout pharmacological treatments in both in vitro and in vivo models
[39][51][39,51]. Likewise, some studies suggest the benefits of enhancing phasic GABA signaling during the chronic phase of stroke in humans
[52][53][52,53]. It has been reported that the phasic GABA signaling is increased in cortical pyramidal neurons during the chronic phase of stroke
[29]. Pharmacological boost of α1 subunit-mediated currents at 3 days post stroke promotes functional recovery by targeting cortical plasticity
[29].
Although glutamate is excitotoxic during the acute phase following stroke, it plays a beneficial role during the recovery phase by inducing LTP
[33][34][33,34]. Indeed, studies in humans have suggested that the stimulation of the penumbra cortex by boosting local excitability as soon as 7 days post stroke improves functional outcome
[54]. However, there is an increase in extrasynaptic levels of GABA due to the reduction in the amount of astrogial GABA transporters on day 7 post stroke in mice
[28]. This event hyperpolarizes neurons at the penumbra area and negatively modulates the induction of LTP
[32][34][55][32,34,55]. Indeed, a recent study using magnetic resonance spectroscopy showed that patients with a low excitatory–inhibitory ratio post stroke had a worse motor outcome
[56]. The application of pharmacological treatments negatively targeting either all α subunits or only α5 subunit-mediated tonic GABA currents at 3 days post stroke has shown significant behavioral recovery in mouse models
[28][34][37][28,34,37]. Interestingly, it has been reported that there is a possible role of extrasynaptic GABA
C receptors, a well-known subclass of GABA
A receptors, in these increased tonic currents during post-acute and chronic phases. The application of antagonists targeting GABA
C receptors from day 3 post stroke improved the motor function of injured mice
[38]. These results together suggest that the time window for the administration of an extrasynaptic GABA
A receptor blocker without affecting its initial neuroprotective role is around 3 days after the infarct, at least in mice.
Overall, the potentiation of symmetric signaling immediately after ischemic stroke counteracts the prominent excitatory cellular state promoting neuronal survival. Based on murine models, the acute phase lasts 3 days, and one of the most important questions to be solved is the exact duration of this phase in humans in order to replicate the results from animal models to patients. In contrast, during the postacute and chronic phase, the rise in tonic GABAergic signaling has to be blocked in order to achieve a better functional outcome. Curiously, the potentiation of phasic inhibition is beneficial during the recovery state. It would be interesting to combine pro-GABA drugs during the acute phase and then change them progressively to both prophasic signaling and antitonic currents.
2.2. Cation–Chloride Cotransporters
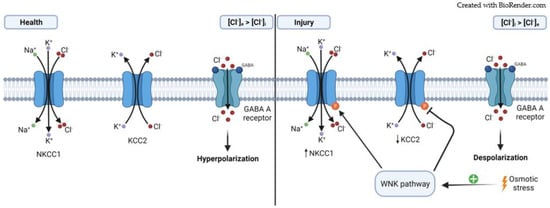
Ionotropic GABAergic signaling is primarily supported by the chloride ion gradient across the plasmatic membrane
[57]. In mature neurons, the regular GABA
A-mediated transmission leads to hyperpolarization by allowing the entry of Cl
− ions that increases the intracellular concentration of chloride ([Cl
−]
i) (
Figure 2)
[26][57][26,57]. The maintenance of [Cl
−]
i mainly depends on cation–chloride cotransporters, where Na
+-K
+-2Cl
− cotransporters (NKCCs) and K
+-Cl
− cotransporters (KCCs) are the most important in the CNS
[26][58][26,58]. The isoform NKCC1 is the only one expressed in the CNS, and its function is to increase the [Cl
−]
i. In contrast, the isoform KCC2 is the main one responsible for decreasing [Cl
−]
i in mature neurons
[26][58][26,58].
Figure 2. Role of NKCC1 and KCC2 cotransporters in the effect of Cl− flow through GABAA receptor. (Health). In a normal situation, the expression of both cotransporters leads to a higher [Cl−]e compared to the [Cl−]i. This results in neuronal hyperpolarization when GABAA receptors are activated; (Injury) After damage, there is an osmotic stress that activates the WNK pathway. This ultimately ends in the phosphorylation of both cotransporters with different outcomes, the expression of KCC2 decreases, whereas the expression of NKCC1 increases; leading to a higher [Cl−]i compared to the [Cl−]e, which results in depolarizing GABAA receptor-mediated responses.
It has been well-documented that under some pathological conditions, [Cl
−]
i can be dysregulated, leading to a depolarizating effect mediated by GABA
A receptors (
Figure 2)
[59]. This is primarily motivated by a high expression of NKCC1 and a low expression of KCC2, leading to a higher [Cl
−]
i compared to the extracellular concentration of chloride ([Cl
−]
e)
[57][58][60][57,58,60]. The main cellular cascade involved in the regulation of NKCC1 and KCC2 following osmotic stress (low [Cl
−]
i) is the With-No-Lysine (K) (WNK) pathway, which ultimately phosphorylates both NKCC1 and KCC2 with opposite outcomes: NKCC1 is activated whereas KCC2 is inhibited (
Figure 2)
[61][62][61,62].
There is compelling evidence showing an increase in neuronal NKCC1 expression immediately after ischemic stroke, an event that contributes to cellular hyperexcitability and cell death
[45][60][63][64][65][66][45,60,63,64,65,66]. Wang and colleagues
[60] have shown that NKCC1 is significantly upregulated in cortical neurons from 3 h to 48 h following focal cerebral ischemia in a rat model. Acute pharmacological treatment using bumetanide, an NKCC1 inhibitor drug, revealed a neuroprotective effect by increasing neuronal survival in an in vitro model of stroke
[45]. This is concordance with previous in vivo results showing a reduction in the infarction volume as well as in the ischemic necrotic cell death, especially remarkable when bumetanide was applied preinjury
[60][63][67][68][60,63,67,68]. A recent study has shown that the inhibition of NKCC1 from day 7 post stroke enhanced axonal sprouting from uninjured neurons, resulting in a significant behavioral improvement
[66].
As previously shown, stroke triggers the WNK signaling pathway leading to both the activation of NKCC1 and the inhibition of KCC2 via phosphorylation (
Figure 2)
[62][69][62,69]. Indeed, the activation of the WNK signaling pathway significantly increased the activity of NKCC1 in cortical and striatal neurons at 6 and 24 h after ischemic stroke in mice
[65]. These findings suggest that blocking the activation of the WNK cascade offers a new therapeutic target to improve the outcome following stroke by targeting NKCC1 activation
[62][69][70][71][72][62,69,70,71,72].
Contrary to what it was seen for NKCC1, the amount of KCC2 at both mRNA and protein levels was downregulated in both rat and mouse models of ischemic stroke
[45][64][66][73][74][45,64,66,73,74]. Curiously, whereas the KCC2 levels in the plasma membrane are notably reduced 3 h post ischemia
[74], there is a progressive decrease in the levels of total KCC2 given that it is significant on days 1 and 7 post stroke, but not at 2 h after injury
[64]. Therefore, a relationship between a maintained expression of KCC2 overtime and the long-term survival rate of neurons has been proposed
[64], which was recently supported
[26][74][26,74]. In this study, hippocampal pyramidal neurons had regular levels of KCC2 and did not display damage signals at 6 h post stroke, but they started to degenerate when KCC2 levels decreased at 48 h after stroke
[26]. In a similar way, an acute blockage of upstream pathways inhibiting KCC2 showed an increased neuronal survival following an ischemic incident in mice
[74]. Therefore, all these evidences seem to point at the upregulation of KCC2 as a therapeutic target to provide protection against stroke-induced cell death. However, similar to the manipulation of GABA signaling, it is a challenge to decipher the timing between acute and recovery phase in humans, and hence, to find the correct point at which to change KCC2 expression/activity from increased to decreased in order to achieve further functional outcome after ischemic stroke
[26].
In summary, blocking NKCC1 during the first hours post stroke has a remarkable effect on neuronal outcome by reducing necrotic death. Likewise, increasing KCC2 levels displays a beneficial role at least during the acute phase, which raises the interesting question of what would happen if KCC2 was manipulated during the post-acute/chronic phase, since higher levels of KCC2 would induce GABA-mediated hyperpolarization leading to a tonic currents-like effect.