Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Rahaba Marima | + 1949 word(s) | 1949 | 2021-12-06 01:54:31 | | | |
| 2 | Camila Xu | Meta information modification | 1949 | 2021-12-10 05:05:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Marima, R. Normal and Aberrant MiRNA and Alternative Splicing Events. Encyclopedia. Available online: https://encyclopedia.pub/entry/16911 (accessed on 28 July 2026).
Marima R. Normal and Aberrant MiRNA and Alternative Splicing Events. Encyclopedia. Available at: https://encyclopedia.pub/entry/16911. Accessed July 28, 2026.
Marima, Rahaba. "Normal and Aberrant MiRNA and Alternative Splicing Events" Encyclopedia, https://encyclopedia.pub/entry/16911 (accessed July 28, 2026).
Marima, R. (2021, December 09). Normal and Aberrant MiRNA and Alternative Splicing Events. In Encyclopedia. https://encyclopedia.pub/entry/16911
Marima, Rahaba. "Normal and Aberrant MiRNA and Alternative Splicing Events." Encyclopedia. Web. 09 December, 2021.
Copy Citation
MiRNAs are short non-coding RNAs that play a central role in regulating RNA silencing and gene expression. Alternative splicing increases the diversity of the proteome by producing several different spliced mRNAs from a single gene for translation. MiRNA expression and alternative splicing events are rigorously regulated processes. Dysregulation of miRNA and splicing events promote carcinogenesis and drug resistance in cancers including breast, cervical, prostate, colorectal, ovarian and leukemia. Alternative splicing may change the target mRNA 3′UTR binding site.
MiRNAs
alternative mRNA splicing (AS)
chemoresistance
1. Introduction
Cancers are biologically diverse and heterogenic and pose a major global challenge with increasing incidence, prevalence and mortality particularly in low resource countries [1][2][3]. Cancer drug resistance poses a major challenge in the treatment and management of the disease. This warrants urgent attention to, and targeting of, molecular alterations, such as miRNA regulation and alternative mRNA splicing, which hold potent therapeutic potential. The same type of numerous alterations in molecular mechanisms that can drive the malignant transformation of cells can also drive the development of drug resistance. Molecular events such as alterations in microRNA (miRNA) transcription and aberrant splicing events contribute to drug resistance in many cancers [1]. MiRNAs are short non-coding RNAs (ncRNAs) that function in the regulation of RNA silencing and gene expression [2]. MiRNAs are involved in diverse biological functions such as cell proliferation and differentiation, homeostasis, metabolism, apoptosis, cell death, and can play a role as either tumor suppressors or as oncogenes [3][4][5].
Alternative splicing (AS) contributes to several cellular functions by maintaining the diversity of the proteome, producing several differently spliced mRNA from a single gene for translation [6]. Like the expression of miRNAs, alternative splicing events are also rigorously regulated processes and aberrant splicing events give rise to malignancy and resistance to therapy [6]. Alternative splicing is known to take place in many genes that can promote or inhibit drug resistance. Cancer cells are known to make use of alternative splicing to alter their sensitivity to chemo-, radio-, hormonal, and immunotherapies by changing the expression profile of various protein isoforms. The changes in alternative splicing and the resulting change in protein expression may affect the effectiveness of various treatments by altering drug uptake and efflux, altering the target of the drug, altering the conversion of the drug to its active state or metabolites, increased drug sequestration, drug inactivation, altered apoptosis, altered DNA damage response (DDR), altered cellular communication and immune system evasion [7].
Evidence suggests that there is an interplay between miRNAs and alternative splicing events that alters drug resistance [1]. However, this interplay is underreported and forms a significant research gap. Dysregulation of miRNA and splicing events promote carcinogenesis and drug resistance in cancers including breast, cervical, prostate, colorectal, ovarian and leukemia [1][4][8]. The increasing prevalence of drug resistance in cancer poses a substantial challenge in the management of the disease. As a result, these molecular alterations are highly attractive drug targets to reverse the aberrant effects of miRNAs and splicing events that promote malignancy and drug resistance.
2. Normal and Aberrant Biosynthesis of MiRNAs
MiRNAs have been identified as vital components in carcinogenesis, including chemoresistance [9]. The mechanisms of action of miRNA in chemoresistance are emerging as crucial factors in anti-cancer therapeutic research. Despite this, these mechanisms remain to be fully understood. A single miRNA can act as a double-edged oncogenic sword, as either a tumor suppressor or an oncogene [10]. MiRNA can target genes related to drug targeting, transport and detoxification, cell cycle regulation, DNA repair and apoptosis (Figure 1) [1][11][12][13][14][15][16][17][18][19]. This process involves the transcription of miRNA transcripts to primary miRNA (pri-miRNA) in the nucleus by RNA pol II. This is followed by pri-miRNA processing to release the precursor miRNA (pre-mRNA) by the Drosha/DGCR8 complex. The released pre-miRNA is about 70nt in length and has a stem loop structure with a 2nt 3′ overhang [20]). The export of the pre-miRNA from the nucleus to the cytoplasm is facilitated by its binding to the Exp5/Ran-GTP complex. In the cytoplasm, the stem-loop pre-miRNA is processed into double-stranded RNA (dsRNA) by the Dicer/TRBP/PACT complex. The helicase enzyme then unwinds the ds-miRNA into single strands. Of the two miRNA strands, the strand with higher stability at the 5′ end will be degraded, while the strand with lower stability at the 5′ end will become a mature miRNA by integrating into the RNA-induced silencing complex (RISC). The miRNA–RISC complex will then bind to the 3′UTR of the target mRNA, inhibiting its translation [20].
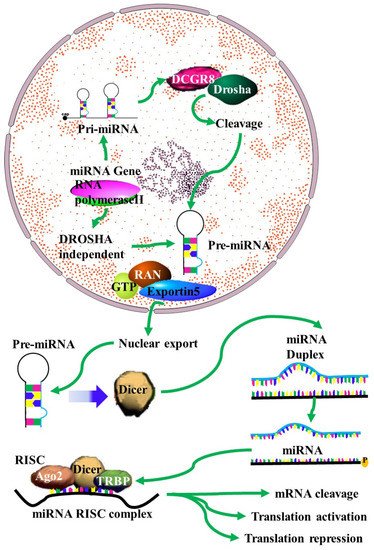
Figure 1. MiRNA biogenesis: A schematic outline of the biogenesis of miRNA. Following transcription by RNA polymerase II, the resulting primary microRNA precursor is then cleaved by the Drosha complex to generate pre-miRNA that is exported to the cytoplasm by Exportin-5. In the cytoplasm it is processed by Dicer into a miRNA duplex. The guide strand (mature miRNA) is then incorporated into the miRNA-induced silencing complex (miRISC) complex where gene silencing can be accomplished via mRNA target cleavage (degradation), or through the prevention of translation [4].
Changes in the levels of miRNA in cancer are largely due to changes in the miRNA biosynthesis pathway. One of the causes of this is changes in the expression of proteins involved in these pathways. For instance, both DROSHA and DICER are downregulated in many cancers [21][22]. However, different oncoproteins have different effects on DROCHA transcription. It is downregulated by ADARB1 [23] but upregulated by MYC [24]. This leads to increased miRNA expression in some cancers and decreased miRNA expression in others. DICER levels in cancer are regulated in part by changes in the transcription factor Tap63, which is responsible for DICER transcription [25]. Mutations in p53 can also lead to a decrease in DICER levels [26], while DICER mRNA is also a target of miRNAs whose expression changes in many cancers [26].
Other than changes in biogenesis, miRNA expression profiles can change as a result of DNA damage response pathways altering the phosphorylation of KH-type splicing regulatory protein. Since some miRNAs can be generated from excised RNA originating from alternate splicing, this can change the expression levels of these miRNAs [27]. Additionally, the export of precursor miRNA to the cytoplasm can result from mutations or altered transcription of the XPO5 gene [28].
3. Normal and Aberrant mRNA Splicing Events: Targeting the 3′UTR
Alternatively spliced mRNA transcripts encode structurally, and perhaps functionally, distinct protein isoforms by exon retention or exclusion. More than 95% of human genes undergo AS. On average human genes have about 8 to 10 exons separated by the non-coding intronic regions, which can be 10 to 100 times longer [6]. In human cells, different mechanistic types of AS exist and these include: (1) intron retention in the mature mRNA transcript; (2) exon skipping where the whole exon is excluded from the mature mRNA transcript, (3) alternative 5′ splice site, with an alternative selection of 5′ splice sites, (4) alternative 3′ splice site, where there is an alternative selection of 3′ splice site. Alternative 5′ or 3′ splice site selection may lead to smaller exon retention, (5) mutually exclusive exons, where distinct exon combinations are generated, (6) alternative promoter selection, where alternative 5′ ends are generated by different pol II promoter selection, (7) alternative polyadenylation sites, where alternative 3′ ends are generated by the selection of different polyadenylation sites [6]. Alternative mRNA splicing is important in normal physiology, but aberrant splicing events have been reported in regard to cancer and drug resistance. It has been reported that aberrant splicing events result in novel mRNA transcripts not observed in normal cells. Furthermore, these novel mRNA transcripts have been reported in cancer and drug resistance [29]. Changes in alternate splicing that occur as a result of cancer largely occur due to genomic splice site point mutations. For instance, multiple splice-site mutations occur in p53 in various types of cancer. Exons are normally flanked by 5′ and 3′ intronic dinucleotides and mutations in these splice sites can lead to exon exclusion [30], or even double exon skipping [31]. Another common mechanism whereby mutations that arise during cancer give rise to aberrant splicing is through the creation of cryptic splice sites. This can lead to the inclusion or exclusion of nucleotides from an mRNA [32]. Cancer has also been observed to have higher rates of mutation in exonic splicing enhancers. Splicing factors bind to these sequences and recruit the spliceosome to splice sites which would normally not be recognized as splice sites. These mutations are known to disrupt the binding of splicing factors such as SC35 and ASF/SF2 [33][34][35].
Mutations in the genes that code for those aforementioned splicing factors that control and regulate splicing also regularly occur in cancers, resulting in changes in alternate splicing. For instance, mutations in the SF3B1 gene are clustered in multiple hotspots. These mutations are all clustered within the Huntingtin, Elongation factor 3, protein phosphatase 2A (HEAT) domain, which is normally associated with intracellular transport [36]. Altered availability of this protein in the cytoplasm may explain the effect these mutations have on the function of these proteins. Mutations in the SRSF2 splicing factor mostly consist of insertions/deletions in a hotspot around residue P95. This region occurs in a linker sequence between the N-terminal RNA recognition motif (RRM) and the C-terminal RS domain. The effect of these mutations is largely negative, being associated with decreased patient survival and poorer treatment outcomes [37]. Mutations in the zinc finger RNA binding motif and serine rich 2 (ZRSR2) splicing factor do not occur in a hotspot and are found across the gene sequence. These mutations mostly lead to reading frame changes and mutant proteins with decreased or no function. This results in altered splicing of some proteins which can best be described as U12-type intron retention [38]. Mutations in the zinc finger RNA binding motif and serine rich 2 (ZRSR2) splicing factor do not occur in a hotspot and are found across the gene sequence. These mutations mostly lead to reading frame changes and mutant proteins with decreased or no function. This results in altered splicing of some protein which can best be described as U12-type intron retention [38].
This results in protein isoforms that may lack full activity, have no activity, or even have the opposite activity. The expression of these isoforms can therefore result in the decreased activity of proteins that function as tumor suppressor genes.
4. MiRNAs Targeting the 3′UTR of Different Transcripts Generated by Alternative Splicing
The ability of miRNAs to bind and regulate mRNA predominantly occurs at the 3′ UTR of the mRNA. Although occasionally this can occur at the 5′ UTR of the target mRNA [39][40]. Several studies have shown that introns retained at the 3′ UTR following alternative splicing can increase the number of putative miRNA target sequences [41], while truncated 3′ UTRs transcripts can alter miRNA binding affinity to targeted mRNA (Figure 2) [42]. The generation of alternative 3′ UTRs occurs through the alternative polyadenylation sites splicing mechanism. The patterns and occurrence of multiple alternative polyadenylation (APA) sites are known to be altered in cancer cells [43]. Ceramide is important for multiple physiological roles and is involved in the actions of some chemotherapy drugs. As a result of this, the downregulation of ceramide synthetase (CerS) is important in acquired drug resistance and metastasis. CerS is downregulated through the interaction between histone deacetylases (HDACs) and miRNAs. In addition to this, an isoform of CerS, CerS1, is detected at higher levels in cancer cells and the CerS1 spliced isoform is targeted for degradation by miR-574-5p by binding to the 3′ UTR within the retained intron between exons 6 and 7 [44].
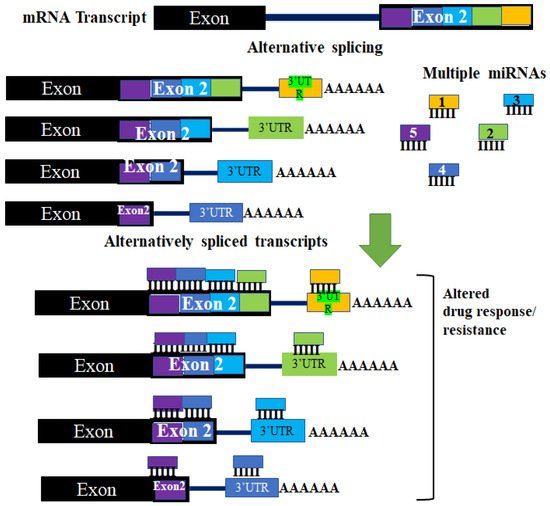
Figure 2. The effect of aberrant alternative splicing in miRNA regulation. Changes in the 3′UTR of target mRNA occur due to different polyadenylation sites. The different 3′ UTRs that result have different regions where miRNAs can bind and target the mRNA. These changes can result in some miRNAs not being able to regulate different alternatively spliced transcripts of the same pre-mRNA. These changes can affect regulation by miRNAs. This may lead to changes in cellular effectors (such as proteins and enzymes) of drug resistance, thereby altering a tumor’s response to a drug.
References
- Siegfried, Z.; Karni, R. The role of alternative splicing in cancer drug resistance. Curr. Opin. Genet. Dev. 2018, 48, 16–21.
- Gebert, L.F.R.; MacRae, I.J. Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 2019, 20, 21–37.
- Ma, J.; Dong, C.; Ji, C. MicroRNA and drug resistance. Cancer Gene Ther. 2010, 17, 523–531.
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenet. 2019, 11, 25.
- Zhang, Y.; Wang, J. MicroRNAs are important regulators of drug resistance in colorectal cancer. Biol. Chem. 2017, 398, 929–938.
- Wang, B.D.; Lee, N.H. Aberrant RNA Splicing in Cancer and Drug Resistance. Cancers 2018, 10, 458.
- Sciarrillo, R.; Wojtuszkiewicz, A.; Assaraf, Y.G.; Jansen, G.; Kaspers, G.J.L.; Giovannetti, E.; Cloos, J. The role of alternative splicing in cancer: From oncogenesis to drug resistance. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2020, 53, 100728.
- Si, W.; Shen, J.; Du, C.; Chen, D.; Gu, X.; Li, C.; Yao, M.; Pan, J.; Cheng, J.; Jiang, D.; et al. A miR-20a/MAPK1/c-Myc regulatory feedback loop regulates breast carcinogenesis and chemoresistance. Cell Death Differ. 2018, 25, 406–420.
- Xie, Q.; Wang, S.; Zhao, Y.; Zhang, Z.; Qin, C.; Yang, X. MiR-519d impedes cisplatin-resistance in breast cancer stem cells by down-regulating the expression of MCL-1. Oncotarget 2017, 8, 22003–22013.
- Magee, P.; Shi, L.; Garofalo, M. Role of microRNAs in chemoresistance. Ann. Transl. Med. 2015, 3, 332.
- Pouyanrad, S.; Rahgozar, S.; Ghodousi, E.S. Dysregulation of miR-335-3p, targeted by NEAT1 and MALAT1 long non-coding RNAs, is associated with poor prognosis in childhood acute lymphoblastic leukemia. Gene 2019, 692, 35–43.
- Turrini, E.; Haenisch, S.; Laechelt, S.; Diewock, T.; Bruhn, O.; Cascorbi, I. MicroRNA profiling in K-562 cells under imatinib treatment: Influence of miR-212 and miR-328 on ABCG2 expression. Pharm. Genom. 2012, 22, 198–205.
- Ghodousi, E.S.; Rahgozar, S. MicroRNA-326 and microRNA-200c: Two novel biomarkers for diagnosis and prognosis of pediatric acute lymphoblastic leukemia. J. Cell. Biochem. 2018, 119, 6024–6032.
- Granados-Riveron, J.T.; Aquino-Jarquin, G. Reversal of multidrug resistance of leukemia cells is not necessarily induced by direct miR-138/MDR1 promoter interaction. Leuk. Res. 2017, 57, 55–56.
- Liu, Z.; Hua, Y.; Liu, C.; Cheng, S. MiRNA-494 inhibits apoptosis and promotes autophagy of acute myeloid leukemia cells by downregulating FGFR2. Minerva Endocrinol. 2019, 44, 410–413.
- Wang, L.W.; Wang, H.R.; Ji, W.G.; Guo, S.L.; Li, H.X.; Xu, X.Y. MiRNA-485-5p suppresses the proliferation of acute myeloid leukemia via targeting SALL4. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4842–4849.
- Fan, S.J.; Li, H.B.; Cui, G.; Kong, X.L.; Sun, L.L.; Zhao, Y.Q.; Li, Y.H.; Zhou, J. MiRNA-149 * promotes cell proliferation and suppresses apoptosis by mediating JunB in T-cell acute lymphoblastic leukemia. Leuk. Res. 2016, 41, 62–70.
- Song, B.; Tang, Y.J.; Zhang, W.G.; Wan, C.C.; Chen, Y.; Zhang, L.J. MiR-143 regulates proliferation and apoptosis of myelocytic leukemia cell HL-60 via modulating ERK1. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9237.
- Liu, L.; Ren, W.; Chen, K. MiR-34a Promotes Apoptosis and Inhibits Autophagy by Targeting HMGB1 in Acute Myeloid Leukemia Cells. Cell. Physiol. Biochem. 2017, 41, 1981–1992.
- Vishnoi, A.; Rani, S. MiRNA Biogenesis and Regulation of Diseases: An Overview. Methods Mol. Biol. 2017, 1509, 1–10.
- Merritt, W.M.; Lin, Y.G.; Han, L.Y.; Kamat, A.A.; Spannuth, W.A.; Schmandt, R.; Urbauer, D.; Pennacchio, L.A.; Cheng, J.F.; Nick, A.M.; et al. Dicer, Drosha, and outcomes in patients with ovarian cancer. N. Engl. J. Med. 2008, 359, 2641–2650.
- Karube, Y.; Tanaka, H.; Osada, H.; Tomida, S.; Tatematsu, Y.; Yanagisawa, K.; Yatabe, Y.; Takamizawa, J.; Miyoshi, S.; Mitsudomi, T.; et al. Reduced expression of Dicer associated with poor prognosis in lung cancer patients. Cancer Sci. 2005, 96, 111–115.
- Allegra, D.; Bilan, V.; Garding, A.; Döhner, H.; Stilgenbauer, S.; Kuchenbauer, F.; Mertens, D.; Zucknick, M. Defective DROSHA processing contributes to downregulation of MiR-15/-16 in chronic lymphocytic leukemia. Leukemia 2014, 28, 98–107.
- Wang, X.; Zhao, X.; Gao, P.; Wu, M. c-Myc modulates microRNA processing via the transcriptional regulation of Drosha. Sci. Rep. 2013, 3, 1942.
- Su, X.; Chakravarti, D.; Cho, M.S.; Liu, L.; Gi, Y.J.; Lin, Y.L.; Leung, M.L.; El-Naggar, A.; Creighton, C.J.; Suraokar, M.B.; et al. TAp63 suppresses metastasis through coordinate regulation of Dicer and miRNAs. Nature 2010, 467, 986–990.
- Muller, P.A.; Trinidad, A.G.; Caswell, P.T.; Norman, J.C.; Vousden, K.H. Mutant p53 regulates Dicer through p63-dependent and -independent mechanisms to promote an invasive phenotype. J. Biol. Chem. 2014, 289, 122–132.
- Zhang, X.; Wan, G.; Berger, F.G.; He, X.; Lu, X. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol. Cell 2011, 41, 371–383.
- Melo, S.A.; Moutinho, C.; Ropero, S.; Calin, G.A.; Rossi, S.; Spizzo, R.; Fernandez, A.F.; Davalos, V.; Villanueva, A.; Montoya, G.; et al. A genetic defect in exportin-5 traps precursor microRNAs in the nucleus of cancer cells. Cancer Cell 2010, 18, 303–315.
- Kumar, S.; Kushwaha, P.P.; Gupta, S.J.C.D.R. Emerging targets in cancer drug resistance. Cancer Drug Resist. 2019, 2, 161–177.
- Holmila, R.; Fouquet, C.; Cadranel, J.; Zalcman, G.; Soussi, T. Splice mutations in the p53 gene: Case report and review of the literature. Hum. Mutat. 2003, 21, 101–102.
- Tanko, Q.; Franklin, B.; Lynch, H.; Knezetic, J. A hMLH1 genomic mutation and associated novel mRNA defects in a hereditary non-polyposis colorectal cancer family. Mutat. Res. 2002, 503, 37–42.
- Hoffman, J.D.; Hallam, S.E.; Venne, V.L.; Lyon, E.; Ward, K. Implications of a novel cryptic splice site in the BRCA1 gene. Am. J. Med. Genet. 1998, 80, 140–144.
- Black, D.L. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 2003, 72, 291–336.
- Maniatis, T.; Tasic, B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature 2002, 418, 236–243.
- Colapietro, P.; Gervasini, C.; Natacci, F.; Rossi, L.; Riva, P.; Larizza, L. NF1 exon 7 skipping and sequence alterations in exonic splice enhancers (ESEs) in a neurofibromatosis 1 patient. Hum. Genet. 2003, 113, 551–554.
- Damm, F.; Thol, F.; Kosmider, O.; Kade, S.; Löffeld, P.; Dreyfus, F.; Stamatoullas-Bastard, A.; Tanguy-Schmidt, A.; Beyne-Rauzy, O.; de Botton, S.; et al. SF3B1 mutations in myelodysplastic syndromes: Clinical associations and prognostic implications. Leukemia 2012, 26, 1137–1140.
- Yoshida, K.; Ogawa, S. Splicing factor mutations and cancer. Wiley Interdiscip. Rev. RNA 2014, 5, 445–459.
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat. Commun. 2015, 6, 6042.
- Hinnebusch, A.G.; Ivanov, I.P.; Sonenberg, N. Translational control by 5’-untranslated regions of eukaryotic mRNAs. Science 2016, 352, 1413–1416.
- Baek, D.; Villén, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71.
- Tan, S.; Guo, J.; Huang, Q.; Chen, X.; Li-Ling, J.; Li, Q.; Ma, F. Retained introns increase putative microRNA targets within 3’ UTRs of human mRNA. FEBS Lett. 2007, 581, 1081–1086.
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3’ untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647.
- Ji, Z.; Tian, B. Reprogramming of 3’ untranslated regions of mRNAs by alternative polyadenylation in generation of pluripotent stem cells from different cell types. PLoS ONE 2009, 4, e8419.
- Meyers-Needham, M.; Ponnusamy, S.; Gencer, S.; Jiang, W.; Thomas, R.J.; Senkal, C.E.; Ogretmen, B. Concerted functions of HDAC1 and microRNA-574-5p repress alternatively spliced ceramide synthase 1 expression in human cancer cells. EMBO Mol. Med. 2012, 4, 78–92.
More
Information
Subjects:
Oncology; Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
784
Revisions:
2 times
(View History)
Update Date:
10 Dec 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No